 Allgemeine und Anorganische Chemie (AC1) Allgemeine und Anorganische Chemie (AC1)
Allgemeine und Anorganische Chemie (AC1) Allgemeine und Anorganische Chemie (AC1) Die Versuche in dieser Experimentalvorlesung werden von Georg Monsch, M.Sc., und Dr. Magdalena Rusan vorgeführt.
Di, Mi, Fr, 8:45–10:00 Uhr, Liebig-HS.
Hinweise zur Klausur finden Sie auf der Homepage von Herrn Kornath, wo Sie auch Links zu alten Klausuren finden.
Beachten Sie die Regelung für das Bestehen der Klausur.
• Studierende des Bachelorstudiengangs Chemie und Biochemie,
• Studierende der Biologie,
• Studierende von Lehramtsstudiengängen mit Teilfach Chemie,
• Studierende der Pharmazie (Staatsexamen und Bachelor).
Der Vorlesung Allgemeine und Anorganische Chemie stehen ca. 70 Stunden zur Verfügung. Diese Zeit verteilt sich auf zwei ungefähr gleich große Blöcke:
In Teil A werden die Grundlagen der allgemeinen und anorganischen Chemie in anderer Abfolge – aber hoffentlich doch schlüssig und zusammenhängend – dargestellt als in den Lehrbüchern üblich. Die Absicht ist, eine optimale Vernetzung mit dem Chemischen Grundpraktikum herzustellen, das von den Studierenden des 1. Semesters des Bachelorstudiengangs Chemie und Biochemie absolviert wird. Mit dem Abschluss von Kapitel 11 Ende November sind alle für das Praktikum wichtigen Begriffe behandelt worden. Anschließend werden diese Grundlagen im dann folgenden systematischen Teil benutzt, um Ordnung in die Vielfalt der stofflichen Welt zu bringen. Für die Klausur am Ende der Vorlesung können Sie den Stoff zusammenhängend aus einem der Lehrbücher vertiefen, da in der Vorlesung nur die Abfolge, aber nicht der typische Umfang des Stoffes verändert wurde.
Für Studierende im Bachelorstudiengang Chemie und Biochemie sowie für Studierende der Biologie und des Lehramtes wird eine Übung von Prof. Kornath (1 SWS) angeboten. Herr Kornath entwirft für diese Übung Aufgabenblätter, die Sie unter Anleitung einer Tutorin/eines Tutors bearbeiten.
Für die Hörer aus dem Staatsexamensstudiengang Pharmazie ist keine Übung vorgesehen, da hier die Vorlesung das anorganisch-analytische Praktikum begleitet, in dessen Rahmen eigene Seminare abgehalten werden.
Parallel zu dieser Vorlesung findet im ersten Semester des Bachelorstudiengangs Chemie und Biochemie das Liebig-Lab statt, das von Dr. Böttcher organisiert wird. Dieses Praktikum wird von der Vorlesung zum Chemischen Grundpraktikum von Dr. Böttcher begleitet, dessen Inhalt mit der Grundvorlesung abgestimmt ist. Zusammen bilden (1) diese Grundvorlesung, (2) das Chemische Grundpraktikum und (3) die Vorlesung zum Chemischen Grundpraktikum Ihre Ausbildung in Allgemeiner und Anorganischer Chemie im 1. Semester.
Anschließend an diese Vorlesung finden das Chemische Praktikum für Biologen und das Anorganisch-chemische Praktikum I für Lehramtstudierende statt. Bei diesen Praktika wird das Bestehen der Klausur zu dieser Vorlesung als Eingangsvoraussetzung verlangt. Hinweise auf diese Praktika finden Sie daher oben im Klausur-Abschnitt.
Lehrbücher, aus denen der Stoff im Zusammenhang gelernt werden kann:
C. E. Mortimer, U. Müller: Chemie. 10. Auflage, Thieme 2010 (ISBN 978-3-13-484310-1).
E. Riedel, C. Janiak: Anorganische Chemie. 8. Auflage, de Gruyter 2012, ISBN 978-3-11-022566-2. Die Stoffchemie ist ausgewogen dargestellt, das heißt, umfangreicher als im Mortimer, aber nicht so enzyklopädisch wie in den umfangreicheren Gesamtdarstellungen. Ein Problem, das auch beim Mortimer auftritt, ist die Einhaltung der Oktettregel. Wir werden sie in der Vorlesung strikt beachten, in beiden Büchern wird das auch so angesagt, aber nicht konsequent beibehalten. Wir werden das Thema in der Vorlesung ansprechen.
M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie. 2. Auflage, Spektrum Akademischer Verlag 2010, ISBN 978-3-8274-2533-1
Umfangreichere Gesamtdarstellungen:
Holleman, Wiberg: Lehrbuch der Anorganischen Chemie. Walter de Gruyter 2007, ISBN 978-3-11-017770-1
C. E. Housecroft, A. G. Sharpe: Anorganische Chemie. Pearson 2006, ISBN 3-8273-7192-9
Atkins, Overton, Rourke, Weller, Armstrong: Inorganic Chemistry. 5th ed., Oxford University Press 2010, ISBN 978-0-19-923617-6
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Zu den häufig benutzten Sonderzeichen gehören griechische Buchstaben: α (alpha), β (beta), π (pi), σ (sigma), χ (chi). Wie Sie sehen, werden α und π durch die verwendete Schrift (Verdana) nicht so deutlich dargestellt wie zum Beispiel durch Schriften aus der Helvetica- (α, π) oder Times-Roman-Familie (α, π). Wenn das beim Lernen nervt, sagen Sie bitte Bescheid.
Diese Website soll (1) die Gliederung der Vorlesung zeigen, (2) Abbildungen zur Verfügung stellen, (3) Organisatorisches übersichtlich an einer Stelle zusammenfassen, (4) ausgearbeiteten Text da zeigen, wo die empfohlenen Lehrbüchern ergänzt werden sollen, meistens handelt es sich dabei um das jeweilige konkrete Beispiel, mit dem ein Sachverhalt erklärt wird. Diese Website soll nicht – ein Lehrbuch ersetzen. Im Gegenteil – Sie werden zahlreiche Querverweise zu den Lehrbüchern finden.
Wenn Sie das Skript oder Teile davon ausdrucken möchten, verwenden Sie am Besten die pdf-Version (Stand: 12. Oktober 2016).
Erläuterungen zu den unterschiedlichsten Themen, die nicht so recht in den Haupttext passen, sind alle in einem Dokument gesammelt. Ein weiteres Dokument enthält Vereinbarungen der Dozenten der Veranstaltungen des Lehrbereichs AC für das 1. Semester.
Chemie ist die Lehre von den Stoffen und den Stoffumwandlungen. Um Stoffe aus der natürlichen Umgebung in ihrer Funktion zu verstehen – oft auch um sie für eine Anwendung nutzbar zu machen, ist die Chemie häufig einem bewährtem Vorgehen gefolgt, das in der Regel mehrfach durchlaufen wird: Der Analyse von Stoffen aus Natur und Technik folgt die Synthese neuer Stoffe, die zum Verständnis der Natur beitragen oder die für eine bestimmte Anwendung optimiert sind. Schwerpunkte angewandter chemischer Forschung sind die Wirkstoffsynthese und die Werkstoffsynthese. Aus diesen beiden Bereichen soll jeweils ein Beispiel das genannte Prinzip veranschaulichen.
Schon früh wurde ein Extrakt des Opiums, dem eingetrockneten Milchsaft aus den unreifen Samenkapseln von Schlafmohn (Papaver somniferum L.) als Schmerzmittel eingesetzt, wobei dessen hohes Suchtpotential als wichtigste Nebenwirkung in Kauf zu nehmen war. Die Analyse des Extrakts ergab, dass die schmerzstillende Wirkung auf Morphin als Hauptwirkstoff zurückzuführen ist (Sertürner 1806). Die daraufhin eingeführte Verwendung von reinem Morphin milderte zwar einige lästige, aber zweitrangige Nebenwirkungen des Gesamtextrakts (zum Beispiel die hartnäckige Verstopfung), nicht aber die suchterzeugende Wirkung des Opiums, für die vor allem Morphin selbst verantwortlich ist. Die Aufklärung der Morphinstruktur war anschließend die Grundlage einer chemischen Modifizierung des Wirkstoffmoleküls, bei der tatsächlich Stoffe synthetisiert wurden, die bei annähernd gleicher schmerzstillender Aktivität weniger suchterzeugend sind (Opiat-Analgetika wie Pethidin und Methadon). Die Entwicklung verlief nicht ohne schwerwiegende Irrtümer: Heroin verdankt seinen Namen der verfrühten Freude seines Erfinders, der die Heldentat vollbracht zu haben glaubte, den gesuchten Morphin-Ersatzstoff ohne Suchtpotential entdeckt zu haben.
Einige Zeit nach diesen Entwicklungsschritten führte die weitere Analyse der Morphinwirkung zur Entdeckung von körpereigenen Opiaten (Endorphinen) mit einer zur Zeit nur erhofften Anwendung in der Zukunft.
Mit dem Beginn der Bronzezeit lernte der Mensch erstmals, Metalle zur Herstellung von Waffen, Werkzeugen und Schmuck zu nutzen. Bronze ist eine Legierung aus Kupfer und Zinn, deren Herstellung einige Kenntnisse voraussetzt. Während nämlich das in Mitteleuropa verhältnismäßig häufige Gold gediegen vorkommt und daher „nur“ in hinreichender Menge gesammelt und zu Münzen oder Schmuckstücken wie den schweren keltischen Halsringen geschmolzen werden musste, kommt Kupfer selten und Zinn nie gediegen vor. Das Hauptvorkommen von Zinn ist vielmehr Zinndioxid in Form des Minerals Cassiterit. Es musste also die Exploration von Erzlagerstätten und die Verhüttung des Erzes beherrscht werden, um aus „Steinen“ ein Metall zu gewinnen.
In der nachfolgenden Eisenzeit gelang es, das in viel größerer Menge verfügbare Eisen nutzbar zu machen, wozu allerdings höherere Anforderungen an die Verhüttungstechnologie zu meistern waren, die im wesentlichen im heute betriebenen Hochofenprozess wiederzufinden sind. So verstanden es frühe Kulturen wie die der Etrusker, die zum Beispiel auf Elba reichlich vorkommenden Eisenoxidminerale Hämatit (siehe obere Abbildung, darunter ein Beispiel für ein früh genutztes Vorkommen in den Ammergauer Alpen) und Magnetit durch Holzkohle zu Eisen umzusetzen. Dabei gibt es genug Hinweise, dass schon die frühen Nutzer von Eisen in der Lage waren, Waffen aus hochwertigem Stahl zu erzeugen, wozu vor allem die Einstellung des korrekten Kohlenstoffgehalts gelingen musste.

Ein ca. 1 m großer Eisenoxidbrocken am Monte Calamita im Südosten Elbas (ital. calamita, Magnet). Das Vorkommen wurde beginnend mit der Etruskerzeit bis zum 2. Weltkrieg ausgebeutet.

Ein einheimisches Eisenerzvorkommen: die wohl seit dem frühen Mittelalter bis ca. 1840 ausgebeutete St.-Mang-Grube beim Säuling (Füssen).
Mit dem Aufkommen moderner werkstoffkundlicher Analysenmethoden konnte der Zusammenhang zwischen dem atomaren Aufbau der Legierungen sowie den Wechselwirkungen zwischen den Atomen und den Legierungseigenschaften erforscht werden. Heute gelingt es auf der Grundlage solcher Methoden, zum Beispiel der Röntgenbeugung, selbst so erstaunliche Eigenschaften zu erklären, wie sie bei den sogenannten „Gedächtnislegierungen“ gefunden werden, die nach einer Verformung beim bloßen Erwärmen ihre ursprüngliche äußere Form wieder einnehmen.
•Versuch: „Thermobil“.
Gemische (Granit, Honig, Luft) können durch Trennverfahren wie Destillation oder Chromatographie in Reinstoffe zerlegt werden. Ein Reinstoff ist durch charakteristische Eigenschaften und Kennzahlen definiert, zum Beispiel durch sein optisches und magnetisches Verhalten. Die Umwandlung von Stoffen ist Gegenstand der Chemie. Verbindungen können durch chemische Reaktionen in Elemente zerlegt werden. Der Aufbau von Verbindungen aus den Elementen folgt den stöchiometrischen Gesetzen, die auf den atomaren Aufbau der Materie hinweisen. Atome als die kleinsten Teilchen eines Elementes bauen in dem durch die Summenformel angegebenen Zahlenverhältnis eine Verbindung auf; mit der konstanten Masse einer Atomsorte liegt damit auch das Masseverhältnis der Elemente in einer Verbindung fest. Das Mol verbindet die atomare Masseeinheit mit der Gramm-Skala. Es erlaubt die Formulierung von Reaktionsgleichungen und das stöchiometrische Rechnen. Für Gase gelten darüberhinaus Volumengesetze, denen der molekulare Aufbau entnommen werden kann. Die Moleküle wichtiger Gase wie Stickstoff, Sauerstoff, Wasserstoff und Chlor sind zweiatomig: N2, O2, H2, Cl2.
| Mortimer: 1, 3, 4, 5, 10 Binnewies: Teile von 8 Riedel: 1.1, 3.1–3.4 |
In der Chemie gängige Trennverfahren sind Destillation, Kristallisation, Filtration und Flotation. Für die Trennung kleiner Stoffmengen werden vor allem chromatographische Verfahren eingesetzt, deren Grundprinzip auf dem „Ausschütteln“ beruht (Stichworte: Verteilungsgleichgewicht, stationäre Phase, mobile Phase).
Versuch 2-5: Ausschütteln von Iod aus Wasser • Iod verteilt sich zwischen Wasser und Benzin, es reichert sich dabei in Benzin, in dem es mit violetter Farbe löslich ist, deutlich stärker an als in Wasser, das Iod mit gelber Farbe löst.
Stoffe, die sich durch solche Trennverfahren nicht weiter zerlegen lassen, sind Reinstoffe. Reinstoffe sind durch physikalische Kennzahlen wie Schmelz- und Siedepunkt, Dichte oder Brechungsindex charakterisiert, die unabhängig von der Herkunft des Reinstoffes immer denselben Zahlenwert haben. Auch charakteristische chemische Eigenschaften wie Brennbarkeit oder brandförderndes Verhalten dienen zur Identifizierung von Reinstoffen. Aus dem Alltag bekannte Reinstoffe sind zum Beispiel zahlreiche Mineralien wie Calcit (Kalkstein), Quarz oder Steinsalz (Kochsalz), aber auch Flüssigkeiten oder Gase wie Wasser oder Sauerstoff.
Versuch 1-1: Pt brennt an Luft nicht, Mg brennt an Luft
Versuch 1-4: Fe reagiert mit Luft
Luft hingegen ist ein Stoffgemisch, das sich in Reinstoffe zerlegen lässt.
Versuch 8-1: Fe zeigt 1/5 O2 in Luft an • mit Essigsäure gewaschene Eisenwolle entzieht der Luft einen Bestandteil; die Reaktion kommt zum Erliegen, wenn ca. 1/5 des eingesetzten Luftvolumens verbraucht ist. Luft ist also ein Gasgemisch, das zu ca. 1/5 aus einer reaktiven Komponente besteht, genauer zu 21 Volumen-% aus Sauerstoff, und zu ca. 4/5 aus einem Rest, der unter diesen Bedingungen nicht reagiert, nämlich 78 Vol.-% Stickstoff und 1 Vol.-% Argon.
Die Zerlegung des homogenen Stoffgemischs Luft gelingt durch fraktionierte Destillation. Der Bestandteil mit dem niedrigsten Siedepunkt ist Stickstoff (−196 °C), der als Kühlmittel genutzt werden kann, um die übrigen Bestandteile aus der Luft auszukondensieren (Siedepunkt von Sauerstoff −183 °C); zuvor zwei Versuche, welche die Eigenschaften von flüssigem Stickstoffs zeigen. Stickstoff und Sauerstoff sind Reinstoffe, die an charakteristischen Eigenschaften erkannt werden können, im Versuch sind dies die Farbe und das Verhalten gegenüber einem Magnetfeld.
Versuch 8-17: Sicherheitsaspekte: Gummischlauch in flüssigem Stickstoff; Druckentwicklung beim Verdampfen.
Versuch 8-13: Leidenfrost-Phänomen
Versuch 8-11: Kondensation von flüssigem Sauerstoff an stickstoffgekühltem Kupferfinger (Video im mp4-Format).
Eine chemische Reaktion, eine Stoffumwandlung, hat dann stattgefunden, wenn Stoffe mit anderen physikalischen Kennzahlen und chemischen Eigenschaften entstehen. Erhitzt man zum Beispiel das Mineral Calcit auf ca. 1000 °C, so entstehen zwei Stoffe: ein farbloses Gas und ein weißer Feststoff, der sich jedoch in seinen Kennzahlen, beispielsweise der Dichte, von Calcit unterscheidet. Calcit ist in weitere Bestandteile zerlegt worden, es ist eine Stoffumwandlung eingetreten.
Versuch 3-2: Erhitzen von Kalkstein • es entsteht Kohlendioxid, das durch „Kalkwasser“ nachgewiesen wird; das zurückbleibende weiße Pulver ist scheinbar unverändert, zeigt aber andere Eigenschaften als das eingesetzte Kalksteinpulver.
Bei dem Versuch, die in der Natur vorliegenden Stoffe in Reinstoffe aufzutrennen und diese dann durch chemische Reaktionen weiter zu zerlegen, wurden chemische Grundstoffe, die chemischen Elemente entdeckt. Dies sind Stoffe, die sich nicht weiter in andere Stoffe zerlegen lassen. Umgekehrt entstehen durch Reaktion der chemischen Elemente miteinander alle übrigen Stoffe, die Verbindungen. In der Natur wurden 92 dieser chemischen Elemente gefunden, denen nahezu 5.000.000 bekannte Verbindungen gegenüberstehen.
Schon vor etwa 200 Jahren wurden Gesetzmäßigkeiten entdeckt, die darauf hindeuten, dass die chemischen Elemente aus gleichartigen, mit einer bestimmten charakteristischen Masse behafteten kleinsten Teilchen, den Atomen, aufgebaut sind. Die erste Regel ist das
• Gesetz von der Erhaltung der Masse, wonach bei allen chemischen Reaktionen die Gesamtmasse der an der Reaktion beteiligten Stoffe konstant bleibt. Bei chemischen Reaktionen erfolgt lediglich eine Umgruppierung dieser Atome, die Gesamtzahl der Atome jeder Atomsorte bleibt unverändert. Außerdem gelten die stöchiometrischen Gesetze:
Versuch 4-2: Fe wird beim Verbrennen schwerer
Versuch 4-3: Fe verbrennt im geschlossenen System ohne Änderung der Gesamtmasse
• Das Gesetz der konstanten Proportionen besagt, dass sich eine chemische Verbindung immer aus konstanten Mengenverhältnissen der Elemente bildet. So verbindet sich 1,000 g des Elementes Kohlenstoff immer mit 1,333 g des Elementes Sauerstoff zur Verbindung „Kohlenmonoxid“, aber nicht mit davon abweichenden Mengen wie 1,350 g oder 1,500 g; eine zweite Verbindung aus Kohlenstoff und Sauerstoff, „Kohlendioxid“, enthält auf 1,000 g Kohlenstoff stets 2,667 g Sauerstoff. 1,00 g des Elementes Wasserstoff verbindet sich stets mit 7,95 g Sauerstoff zur Verbindung Wasser.
Versuch 4-4: Zeigen: PbO, Pb3O4, PbO2
• Das Gesetz der multiplen Proportionen lautet: Bilden wie im Fall von Sauerstoff und Kohlenstoff zwei Elemente mehrere Verbindungen miteinander, dann stehen die Massen der Elemente zueinander im Verhältnis kleiner ganzer Zahlen. So reagiert 1,000 g Kohlenstoff mit 1,333 g Sauerstoff zu Kohlenmonoxid; weiterhin reagiert 1,000 g Kohlenstoff mit 2,666 g Sauerstoff zu Kohlendioxid. Bezogen auf die gleiche Menge Kohlenstoff (1,000 g) stehen die Massen von Sauerstoff im Verhältnis 1,333 : 2,666 = 1 : 2.
Verbindungen entstehen durch chemische Reaktion verschiedener Elemente. So verbrennt Kohlenstoff in Anwesenheit von Sauerstoff zu Kohlenmonoxid und Kohlendioxid (die Symbole „C“ und „O“ stammen von neulat. carboneum und oxygenium; wenn Sie sich für die Herkunft der Elementsymbole und -namen interessieren, so finden Sie im Holleman-Wiberg erschöpfende Information):
2 C + O2 → 2 CO
C + O2 → CO2
CO und CO2 sind die Summenformeln für Kohlenmonoxid und Kohlendioxid. Summenformeln geben die Atomsorten an, aus denen eine Verbindung aufgebaut ist, sowie deren Zahlenverhältnis; bei bekannter Atommasse damit also auch die Massenverhältnisse der in der Verbindung miteinander verbundenen Elemente.
Eine Summenformel enthält keine Information über die Struktur der Verbindung, auch nicht über die Art der Bindung zwischen den Atomen. So sind CO und CO2 Gase, in denen einzelne Moleküle der Formeln CO und CO2 vorliegen, währen SiO2 (Quarz) keine isolierten Moleküle enthält, sondern ein polymerer Stoff ist, in dem n Atome Silicium mit 2 n Atomen Sauerstoff einen Kristall aufbauen. NaCl und Na2CO3 sind aus Ionen aufgebaute Salze, deren Kristalle aus n Na+- und n Cl−-Ionen bzw. aus 2 n Na+- und n CO32−-Ionen aufgebaut sind.
Trotz dieser Einschränkung werden die Formeln von Gasen und Flüssigkeiten so hingeschrieben, dass die Zusammensetzung der charakteristischen kleinsten Teilchen, der Moleküle, wiedergegeben wird. Daher wird Sauerstoff als O2 und nicht als O angegeben, da sich herleiten lässt (siehe Abschnitt „Gase“), dass Sauerstoff unter den normalen Umgebungsbedingungen aus zweiatomigen Molekülen aufgebaut ist.
Die Masse von Atomen und Molekülen kann in jeder beliebigen Masseeinheit angegeben werden. So wiegt ein Sauerstoffatom 2,66 × 10−23 g. Um solch unhandliche Zahlen zu vermeiden, wird als Einheit der Atommasse 1/12 der Masse eines Atoms des Kohlenstoffisotops 12C festgelegt (zum Begriff „Isotop“ siehe unten). Diese Masseeinheit wird mit u oder Dalton bezeichnet oder auch ohne Dimension angegeben. Für die Umrechnung in Gramm gilt:
1 u (= 1 D = 1 atomare Masseneinheit) = 1,6605 × 10−24 g
Die relative Molekülmasse Mr ist die Masse eines Moleküls in Atommasseneinheiten. Sie ist gleich der Summe der Atommassen der im Molekül oder der Summenformel enthaltenen Atome. Besteht eine Verbindung nicht aus einzelnen Molekülen (zum Beispiel eine Ionenverbindung wie Steinsalz, NaCl), so wird der Begriff „Formelmasse“ verwendet. So errechnen sich mit den entsprechenden Atommassen (C 12,01, O 16,00, Na 22,99, Cl 35,45) die Molekülmasse von CO2 und die Formelmasse von NaCl zu:
Mr(CO2) = 12,01 + 2 × 16,00 = 44,01
Mr(NaCl) = 22,99 + 35,45 = 58,44
Ein zentraler Begriff in der Chemie ist das Mol (SI-Symbol: mol). 1 mol ist die Stoffmenge, die aus genau so vielen Teilchen (Atomen, Molekülen, Formeleinheiten) besteht, wie Atome in 12 g 12C enthalten sind. So enthält nicht nur 1 mol 12C, sondern 1 mol eines jeden Stoffes NA = 6,02217 × 1023 Teilchen. NA ist die Avogadrosche Zahl. Die Stoffmenge n(X) eines Stoffes X ergibt sich aus der Masse m(X) der Probe und der molaren Masse M(X) des Stoffes.
Beachte: Die Masse m des Stoffes wird in g angegeben, seine molare Masse M in g mol−1. Der Zahlenwert von M entspricht der relativen Masse Mr des Stoffes. So wiegen Sie die folgenden Massen m an Kohlendioxid, Natriumchlorid, Natrium oder Sauerstoff ab, wenn die Stoffmenge n = 1 mol benötigt wird:
m(CO2) = 1 mol × M(CO2) = 1 mol × 44,01 g mol−1 = 44,01 g
m(NaCl) = 1 mol × M(NaCl) = 1 mol × 58,44 g mol−1 = 58,44 g
m(Na) = 1 mol × M(Na) = 1 mol × 22,99 g mol−1 = 22,99 g
m(O2) = 1 mol × M(O2) = 1 mol × 32,00 g mol−1 = 32,00 g
Ist dagegen von 1 mol Sauerstoffatomen die Rede, so ist deren Masse:
m(O) = 1 mol × M(O) = 1 mol × 16,00 g mol−1 = 16,00 g
Für das ideale Gas gilt: 1 mol nimmt unter Standardbedingungen (1 atm = 1,013 bar = 101,3 kPa, 0 °C) ein Volumen von 22,4 L ein, das heißt, das Molvolumen des idealen Gases beträgt 22,4 L.
Mit Hilfe der relativen Atommassen und der Summenformel einer Verbindung lassen sich die Masseanteile der einzelnen Bestandteile errechnen.
Beispiel: Wieviel Masse-% Aluminium enthält Aluminiumoxid, Al2O3? Mit den gerundeten Atommassen von Al (27) und O (16) ergibt sich für die Formelmasse von Al2O3:
Mr = 2 × 27 + 3 × 16 = 54 + 48 = 102
Der Masseanteil an Al ist dann 54/102 = 0,53, also 53 Masse-%.
Eine umgekehrte Rechnung zeigt, wie man aus dem Ergebnis einer chemischen Analyse, bei der die prozentuale Zusammensetzung einer Verbindung ermittelt wird, eine Summenformel errechnen kann.
Die Frage lautet: Wie ist die Summenformel einer Verbindung mit folgendem Analyseergebnis: Na: 32,85 %, Al: 12,85 %, F: 54,30 %? Bekannt sind die Atommassen: Na = 22,99, Al = 26,98, F = 19,00. Zuerst wird nun durch Division des prozentualen Masseanteils durch die relative Atommasse das Atomzahlverhältnis Na:Al:F bestimmt zu 32,85/22,99 : 12,85/26,98 : 54,30/19,00 = 1,4289:0,4763:2,8579. Division durch den kleinsten Wert (0,4763) führt zu einem ganzzahligen Verhältnis von 3,00:1:6,00, die Summenformel ist also Na3AlF6.
Bei Mengenberechnungen aus chemischen Gleichungen wird der Molbegriff verwendet, durch den die mikroskopische Welt der Atome mit wägbaren Mengen verknüpft wird. Hierzu das folgende Beispiel: Bei der Reaktion von 1 mol Natriumcarbonat (Soda) mit überschüssiger Salzsäure werden 2 mol Säure verbraucht und es entstehen 2 mol Natriumchlorid (Steinsalz) neben 1 mol Kohlendioxid und 1 mol Wasser. Die Reaktionsgleichung zeigt die molaren Mengen:
Na2CO3 + 2 HCl → 2 NaCl + H2O + CO2
Mit den gerundeten Atommassen (Na = 23, C = 12, O = 16, H = 1, Cl = 35,5) ergeben sich die Formelmassen der beteiligten Verbindungen; diese in Gramm genommen ergeben die umgesetzen Mengen für die oben angegebenen Molzahlen:
106 g Na2CO3 + 73 g HCl → 117 g NaCl + 18 g H2O + 44 g CO2
Eine Summenformel kann so dazu dienen, aus der eingesetzen Menge eines Reaktanden die Mengen der übrigen Stoffe zu berechnen. Beispiel: Wieviel Liter CO2 entstehen bei der oben angegebenen Umsetzung aus 1 kg Soda? Antwort:
1 mol Na2CO3 ergibt gerade 1 mol CO2. Bei Normalbedingungen und mit einem Molvolumen von 22,4 L für CO2 ergibt sich: 1000 g/106 g mol−1 × 22,4 L mol−1 = 211 L.
Versuch 4-6: Mol-Würfel (22.4 L) aufstellen
Chemische Reaktionen sind durch einen Stoff- und Energieumsatz gekennzeichnet. Bei den folgenden "exothermen" Reaktionen wird Energie in unterschiedlicher Form freigesetzt.
Versuch 3-1: Eisen + Schwefel • gemäß Fe + S → FeS entsteht Eisen(II)-sulfid, der Nachweis der Stoffumwandlung gelingt leicht durch einen Magneten.
Versuch 3-13: pyrophores Eisen • feinst verteiltes, oberflächenreiches Eisen ist viel reaktiver als kompakte Formen; duch Erhitzen von Eisen(II)-oxalat lässt sich gemäß FeC2O4 → Fe + 2 CO2 elementares Eisen herstellen, das beim bloßen Kontakt mit Luft sofort entflammt.
Versuch 3-3: Kaliumchlorat + Schwefel • die Reaktion 2 KClO3 + 3 S → 2 KCl + 3 SO2 verläuft so heftig, dass nur mit kleinsten Mengen experimentiert wird, um es schön knallen zu lassen.
Versuch 3-5: Ammoniumdichromat-Vulkan • gemäß (NH4)2Cr2O7 → Cr2O3 + N2 + 4 H2O entsteht in einer prächtig anzuschauenden Reaktion ein Chrom(III)-oxid-Krater.
Versuch 3-7: Zn + H2 in Pt-Schale • ein besonders interessantes Experiment; dieser einfache Versuchsaufbau reicht aus, um elektrischen Strom zu erzeugen: eine Zinkstange wird in eine Platinschale getaucht, die mit Salzsäure gefüllt ist. Zinkstange und Platinschale sind durch einen Draht mit einem Voltmeter verbunden, dass etwas mehr als 0,7 V Spannung anzeigt (bei dem Versuch darf das Zink das Platin nicht berühren!). Versuchen Sie spätestens nach dem Kapitel „Elektrochemie“, diesen Versuch zu erklären.
Versuch 3-8: Luminol • eher seltener ist die Freisetzung von Lichtenergie bei einer chemischen Reaktion. Aus der Natur ist dieses Phänomen von Glühwürmchen bekannt, die ebenso wie bei dem hier gezeigten Versuch ein „kaltes“ Leuchten erzeugen können. Wir werden vor allem beim Versuch „Singulettsauerstoff“ auf die Grundlagen eingehen.
Reaktionen, die unter Energieverbrauch ablaufen, sind endotherm:
Versuch 3-12: Bariumhydroxid und Ammoniumthiocyanat.
Bei einer chemischen Reaktion findet eine Umverteilung von Atomen statt. Die Versuche zeigen, dass dabei neben der stofflichen Veränderung auch Energie umgesetzt wird. Es gibt chemische Reaktionen, bei denen Energie freigesetzt wird und andere, bei denen Energie verbraucht wird. Die bei einer chemischen Reaktion entwickelte oder verbrauchte Wärmemenge heißt Reaktionswärme. Im SI-System werden Reaktionswärmen genau wie andere Energieformen in Joule (J) angegeben (früher: kcal; 1 kcal = 4,187 kJ). Die Reaktionswärme einer bei konstantem Druck ablaufenden chemischen Reaktion heißt Reaktionsenthalpie. Das Symbol ist ΔH; sind die angegebenen Zahlenwerte auf Standardbedingungen bezogen (T = 25 °C = 298,15 K; p = 1 atm = 1,013 bar = 101,3 kPa), lautet das Formelzeichen ΔH° (Das Superskript ° ist eins von zwei IUPAC-empfohlenen Symbolen, um den Standardzustand zu kennzeichnen; beim zweiten Symbol kommt ein Querstrich durch das ° hinzu, was dann so aussieht: ΔH⦵). Ein tiefgestellter Buchstabe hinter Δ kann zur weiteren Unterscheidung genutzt werden (r für Reaktion, f für Bildung, vap für Verdampfung, sub für Sublimation, fus für Schmelzen, at für Atomisierung, etc.). Zwei Beispiele:
Bei der Bildung von Ammoniak aus Wasserstoff und Stickstoff gemäß
3/2 H2 + 1/2 N2 → NH3
wird unter Standardbedingungen pro mol Formelumsatz (das heißt, man liest die Reaktionsgleichung in mol) eine Reaktionswärme von 46,2 kJ entwickelt und an die Umgebung abgegeben; die Reaktionsenthalpie beträgt dann −46,2 kJ mol−1, da freiwerdende Energiebeträge ein negatives Vorzeichen erhalten (der Energieinhalt der Endstoffe ist kleiner als derjenige der Ausgangsstoffe). Die gesamte Reaktionsgleichung mit Stoff- und Energiebilanz lautet dann:
3/2 H2 + 1/2 N2 → NH3 ΔrH° = −46,2 kJ mol−1
Will man gebrochene Koeffizienten in der Reaktionsgleichung vermeiden, so werden Stoff- und Energiemengen mit einem entsprechenden Faktor multipliziert, zum Beispiel:
3 H2 + N2 → 2 NH3 ΔrH° = −92,4 kJ mol−1
Bei der Bildung von Stickstoffmonoxid aus Stickstoff und Sauerstoff nach der folgenden Gleichung wird pro mol Formelumsatz eine Reaktionswärme von 90,4 kJ verbraucht, also der Umgebung entzogen. Aus der Umgebung entzogene Wärme erhält ein positives Vorzeichen (der Energieinhalt der Endstoffe ist größer als der der Ausgangsstoffe), die Gleichung lautet dann:
1/2 N2 + 1/2 O2 → NO ΔrH° = 90,4 kJ mol−1
oder
N2 + O2 → 2 NO ΔrH° = 180,8 kJ mol−1
Reaktionen mit negativem ΔH werden exotherm, Reaktionen mit positivem ΔH werden endotherm genannt. Die Reaktionsenthalpie hängt von der Temperatur und vom Druck bei der Reaktion ab. Die bei den Beispielen angegebenen Zahlenwerte beziehen sich daher auf den oben definierten Standardzustand.
Eine chemische Verbindung kann auf verschiedenen Reaktionswegen entstehen. So kann Kohlendioxid, CO2, direkt durch die Verbrennung von Kohlenstoff in Sauerstoff erhalten werden (Weg 1):
C + O2 → CO2 ΔrH° = −393,8 kJ mol−1
Die Verbrennung lässt sich aber auch so steuern, dass anstelle von CO2 Kohlenmonoxid, CO, entsteht; dieses kann dann in einer zweiten Reaktion zu CO2 weiterverbrannt werden (Weg 2):
C + ½ O2 → CO ΔrH° = −110,6 kJ mol−1
und
CO + ½ O2 → CO2 ΔrH° = −283,2 kJ mol−1
Summe der Enthalpiewerte beim Weg 2: ΔrH°ges = −393,8 kJ mol−1
Die Reaktionsenthalpie beim Weg 1 ist der Summe der Enthalpien beim Weg 2 gleich; für Gesamt-Enthalpien gilt allgemein:
ΔrH° (Weg 1) = ΔrH° (Weg 2)
Bei gleichem Anfangs- und Endzustand ergeben verschiedene Reaktionswege also gleiche Reaktionsenthalpien, unabhängig davon, ob eine Reaktion in einem Schritt oder in mehreren Teilschritten durchgeführt wird (Hessscher Satz). Größen wie ΔH, die nur vom erreichten Zustand abhängen, nicht aber vom Weg, auf dem ein System diesen Zustand erreicht hat, heißen Zustandsgrößen.
Der Hesssche Satz erlaubt die Berechnung von Reaktionsenthalpien aus den Reaktionsenthalpien (gedachter) Teilschritte einer Reaktion. Hierzu bedient man sich der sogenannten Standardbildungsenthalpie ΔfH° (Index f von lat. formatio, Bildung, Index ° für Standardbedingungen). Das ist die Enthalpie, die bei der Bildung von 1 mol einer Verbindung aus den Elementen unter Standardbedingungen frei oder verbraucht wird. Ein Beispiel ist die Bildung von NO:
½ N2 + ½ O2 → NO ΔfH° = 90,4 kJ mol−1
Um Standardbildungsenthalpien miteinander vergleichen zu können, setzt man sie für Elemente in ihrem bei Standardbedingungen stabilen Zustand gleich Null. So gilt ΔfH° = 0 für Stickstoff als N2 (nicht N) und Kohlenstoff als Graphit (nicht als Diamant).
Mit Hilfe von Standardbildungsenthalpien und dem Hessschen Satz ist die Berechnung beliebiger Reaktionsenthalpien möglich. Hierzu werden lediglich die Standardbildungsenthalpien der Ausgangsstoffe von den Standardbildungsenthalpien der Endstoffe subtrahiert:
ΔrH° = Σ{ΔfH°(Produkte)} − Σ{ΔfH°(Edukte)}
Beispiel 1: Bildung von „Synthesegas“ aus Kohlenstoff und Wasser (bei den Reaktanden ist der Aggregatzustand angegeben: f = fest, fl = flüssig, g = gasförmig; H2O(g) ist also Wasserdampf bei 25 °C und Normaldruck):
C(f) + H2O(g) → CO(g) + H2(g)
Mit den ΔfH°-Werten für H2O(g) und CO(g) von −241,8 und −110,6 kJ mol−1 ergibt sich:
ΔrH°/kJ mol−1 = −110,6 + 0 − {0 + (−241,8)}
ΔrH° = 131,2 kJ mol−1
Die Reaktion ist also endotherm.
Beispiel 2: Reduktion von Eisen(III)-oxid durch Kohlenmonoxid:
Fe2O3(f) + 3 CO(g) → 2 Fe(f) + 3 CO2(g)
Mit dem ΔrH°-Wert für Fe2O3(f) von −824,8 kJ mol−1 und den oben angegebenen Werten für die Kohlenoxide ergibt sich:
ΔrH°/kJ mol−1 = 2 × 0 + 3 × (−393,8) − {(−824,8) + 3 × (−110,6)}
ΔrH° = −24,8 kJ mol−1
Die Reaktion ist also schwach exotherm.
Dieser Abschnitt vervollständigt die stöchiometrischen Berechnungen. Als Formeln für Gase war bislang nicht eine Summenformel im engeren Sinn verwendet worden, also „O“ für Sauerstoff, sondern O2. Der Nachweis, dass Sauerstoff tatsächlich in Form zweiatomiger Moleküle auftritt, ist ohne Aufwand schnell erbracht, es muss lediglich die Gasdichte bekannt sein, also der Quotient aus Masse und Volumen einer bestimmten Menge Sauerstoff.
Versuch (neu): O2 und CO2 im 2-L-Kolben wiegen
Da der Versuch im Hörsaal nicht unter Standardbedingungen ausgeführt wird – vor allem weil es bei der Messung deutlich wärmer als 0 °C zu sein pflegt, wird zur Auswertung das ideale Gasgesetz genutzt:
p V = n R T
p, V und T sind der Druck, das Volumen und die Temperatur des Gases, n ist wie zuvor die Stoffmenge und R ist die Gaskonstante von 8,31451 kPa L mol−1 K−1. Um zu entscheiden, ob Sauerstoff als O, O2, O3 oder anders zu formulieren ist, soll die molare Masse M bestimmt werden, deren jeweiliger Erwartungswert 16 g, 32 g , 48 g, etc., beträgt. Es gilt:
| M = | m |
| n |
Mit
| n = | p V |
| R T |
gilt:
| M = | m R T |
| p V |
Der in der Vorlesung bestimmte Wert zeigt, dass die Formulierung als O2 korrekt ist.
Die unaufwendige Bestimmung der molaren Masse eines Gases erlaubt auch die Analyse von Stoffen, die sich vollständig zu Gasen zersetzen lassen. Ein Beipiel ist die Bestimmung der Summenformel von Wasser.
Versuch 4-5: Elektrolytische Zersetzung von H2O
Atome sind aus den Nucleonen (Protonen, Neutronen) und Elektronen aufgebaut. Während das Auftreten von Isotopen aus dem Aufbau des Atomkerns verständlich wird, resultieren die chemischen Eigenschaften aus der Elektronenstruktur. Im Mittelpunkt der Betrachtung steht die Beschreibung elektronischer Zustände durch die vier Quantenzahlen n, l, ml und ms. Von diesen wird zuerst die Hauptquantenzahl n eingeführt, mit deren Hilfe sich die Lage der Emissionslinien im Wasserstoffspektrum deuten lassen. Anschließend werden die elektronischen Aufenthaltsräume, die Orbitale, im Detail betrachtet. Die Zuordnung von Elektronenkonfigurationen zu den Grundzuständen der Atome führt zum Periodensystem der Elemente (PSE). Umgekehrt zeigen Atomeigenschaften wie die Ionisierungsenergie oder die Elektronenaffinität einen charakteristischen Gang im PSE.
| Mortimer: 2, 6 Binnewies: 2, 3 Riedel: 1.2, 1.4 |
Die kleinsten Teilchen eines chemischen Elementes, die Atome, sind aus den Elementarteilchen Elektronen, Protonen und Neutronen aufgebaut, die sich in Masse und elektrischer Ladung unterscheiden. Der Atomkern wird durch positiv geladene Protonen der relativen Masse 1,007277 und der Ladung +e sowie durch elektrisch neutrale Neutronen der Masse 1,008665 gebildet. Elektronen sind gegenüber den Kernteilchen, den Nukleonen, nahezu masselos (ca. 1/2000), sie tragen die Ladung −e. 1 e ist die Elementarladung, die kleinste elektrische Ladung; alle auftretenden Ladungsmengen sind ganzzahlige Vielfache dieser Elementarladung. Ihr Betrag ist:
1 e = 1,6022 × 10−19 C
Die Radien der Atomkerne sind von der Größenordnung 10−2 bis 10−3 pm (1 pm = 10−12 m; vergleiche zum Beispiel den Radius eines Kupferatoms von 128 pm, vgl. das Rutherfordsche Experiment). Der im Vergleich zum gesamten Atom sehr kleine Atomkern enthält fast die gesamte Masse des Atoms. Die Zahl der Protonen bestimmt die Größe der positiven Ladung des Kerns, die Kernladungszahl, die der Protonenzahl gleich ist. Die Gesamtzahl der Protonen und Neutronen bestimmt die Masse des Kerns und angenähert die des Atoms, sie wird daher Massenzahl genannt. Ein chemisches Element besteht aus Atomen gleicher Protonenzahl (Kernladungszahl), die Zahl der Neutronen kann jedoch unterschiedlich sein. Die für das Element charakteristische Protonenzahl wird auch Ordnungszahl genannt.
Eine durch Kernladungszahl und Neutronenzahl charakterisierte Atomsorte bezeichnet man als Nuklid. Nuklide mit gleicher Protonenzahl, aber verschiedener Neutronenzahl heißen Isotope. Beispiele sind die Isotope von Wasserstoff (beachte die übliche Schreibweise, die Massenzahl links oben und die Kernladungszahl links unten am Elementsymbol zu notieren): 21H, (= D, Deuterium), 31H (= T, Tritium).
Da die Information über die Ordnungszahl bereits durch das Atomsymbol gegeben wird, werden Nuklide oft nur durch die Massenzahl beschrieben, zum Beispiel bei den verschiedenen Kohlenstoffisotopen:
12C, 13C,14C
oder bei den Isotopen von Stickstoff:
14N, 15N
Die meisten Elemente sind Mischelemente, die aus mehreren Isotopen in oft sehr unterschiedlicher Häufigkeit bestehen. So werden die Isotope von Wasserstoff, Kohlenstoff und Stickstoff mit folgenden Häufigkeiten gefunden: 1H (99,985 %), 2H (0,015 %); 12C (98,89 %), 13C (1,11 %); 14N (99,63 %), 15N (0,36 %).
Einige Elemente sind Reinelemente, die in ihrem natürlichen Vorkommen aus nur einer Nuklidsorte bestehen (zum Beispiel Fluor nur 19F, Natrium nur 23Na). Nuklide mit gleicher Massenzahl, aber unterschiedlicher Kernladungszahl werden Isobare genannt (zum Beispiel 14C, 14N).
Die Untersuchung von Emissionsspektren zeigt eine wesentliche Eigenschaft der Elektronen – die Quantelung der Energiezustände. Atome in einem angeregten elektronischen Zustand können durch Zufuhr thermischer oder elektrischer Energie erzeugt werden. Bei der Rückkehr in den elektronischen Grundzustand wird die Energiedifferenz zum angeregten Zustand oft in Form von Lichtenergie abgestrahlt. Die spektrale Zerlegung des erzeugten Strahlung ergibt Linienspektren als Indiz für diskrete Energiezustände, die von den Elektronen eingenommen werden können.
Versuch 6-1: Li-, Na-, B-Flammenfärbung
Versuch 6-2: Plücker-Röhre (Wasserstoff und Stickstoff) • An einem Glasrohr liegt Hochspannung an. Das Rohr wird mit Wasserstoff gefüllt und anschließend evakuiert. Mit dem Erreichen eines hinreichenden Vakuums beginnt die Röhre blauviolett zu leuchten. Die Leuchterscheinung ist die Mischfarbe der im folgenden Bild dargestellten Spektrallinien.
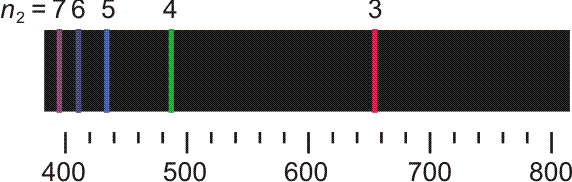
Die ersten fünf Linien der Balmer-Serie. Unter dem Emissionspektrum ist die Wellenlänge in nm angegeben, oberhalb des Spektrums n2, die Hauptquantenzahl des angeregten Zustands. Der energieärmere Zustand liegt bei der Balmer-Serie bei n1 = 2.
Noch bevor eine erklärende Modellvorstellung erkannt wurde, gelang es, eine einfache Beziehung aufzustellen, welche die Lage der Linien in der abgebildeten „Balmer-Serie“ beschreibt: Für n ≥ 3 gilt nämlich (Frequenz ν, Wellenlänge λ):
ν = c λ−1 = 3,289 × 1015 × (1/4 − 1/n2) sec−1
Die Suche nach weiteren Serien in den nicht sichtbaren Bereichen des elektromagnetischen Spektrums führte zu der allgemeinen Beziehung:
ν = 3,289 × 1015 × (1/n12 − 1/n22) sec−1
für n2 ≥ n1. Besonders aufschlussreich ist es, für die Serie mit n1 = 1 – die im UV-Bereich beobachtbare Lyman-Serie – über die beobachteten Linien für n2 = 2, 3, 4, … auf n2 = ∞ zu extrapolieren. Die erhaltene Energie beträgt umgerechnet 13,6 eV. Dieser Betrag entspricht der Ionisierungsenergie des Wasserstoffatoms, das ist die Energie, die für den Vorgang
H → H+ + e−
aufgebracht werden muss (dieser Energiebetrag heißt auch Rydberg-Konstante; Sie finden ihn in den Lehrbüchern auch in anderen Energieeinheiten tabelliert). Es gelang N. Bohr, diese Beobachtungen durch ein Schalenmodell des Wasserstoffatoms zu deuten. Die Elektronen können in diesem Modell nur diskrete Energiezustände einnehmen, deren energieärmster, stabilster Zustand 13,6 eV unter der Ionisationsgrenze liegt, die bei 0 eV gesetzt wird (siehe Abbildung, zusammen mit der Balmer-Serie auch als pdf).
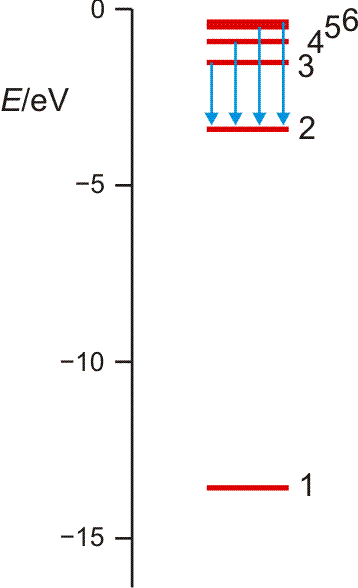
Orbitalenergieschema des Wasserstoffatoms. Die Energie eines Zustands hängt nur von der Hauptquantenzahl n ab, der „Schale“ im Bohrschen Atommodell. Die Ionisationsgrenze liegt beim Ordinatenwert 0. Die blauen Pfeile kennzeichnen die Elektronensprünge, durch welche die Balmer-Serie hervorgerufen wird.
Die erlaubten Zustände treten in immer dichterer Folge auf, je näher die Ionisationsgrenze rückt. Die Energiewerte folgen dabei der Reihe
| E = | 13,6 eV | mit n = 1, 2, 3, … |
| n2 |
n ist eine „Quantenzahl“, mit der eine Elektronenenergie eindeutig beschrieben ist; im Bohrschen Modell wird n als die Nummer der „Schale“ interpretiert, auf der das Elektron um den Kern kreist. Man beachte, dass das Bohrsche Atommodell das Elektron als Teilchen behandelt. So wird die Rydberg-Konstante RH in diesem Modell abgeleitet als (reduzierte Masse μ, Ladung Z, Vakuum-Dielektrizitätskonstante ε0):
| RH = | μ Z2 e4 |
| 8 ε02 h2 |
Es tritt die reduzierte Masse μ auf; für das Wasserstoffnuklid 1H gilt: 1/μ = 1/mProton + 1/mElektron. Die Behandlung des Elektrons als rotierende Masse ist offensichtlich.
Dem Bohrschen Atommodell folgte die wellenmechanische Behandlung der Elektronenstruktur. Eine mathematische Ausarbeitung wird an dieser Stelle nicht gegeben. In Vorlesungen zur Quantenmechanik werden Sie ableiten, dass die „Schale“ des Bohrschen Atommodells nur eine von vier Quantenzahlen darstellt, die den Zustand eines Elektrons eindeutig festlegen. Die ersten drei Quantenzahlen folgen dabei einem hierarchischen Schema, an dessen Ausgangspunkt die Hauptquantenzahl n steht:
Die Hauptquantenzahl kann die Werte n = 1, 2, 3, … annehmen.
Mögliche Werte für die Nebenquantenzahl l sind l = 0, 1, 2, … (n − 1); für l gibt es also n mögliche Werte.
Mögliche Werte für die magnetische oder Orientierungsquantenzahl ml sind ml = −l, …, 0, …, +l; ml hängt damit von l ab, es gibt 2 l + 1 mögliche Werte.
Unabhängig von n, l und ml ist die (magnetische) Spinquantenzahl ms, welche die Werte +½ und −½ annehmen kann.
Man beachte, dass die Hauptquantenzahl n die möglichen Werte für die Nebenquantenzahl l festlegt und diese die möglichen Werte für die Orientierungsquantenzahl ml. Neben der Bezeichnung durch Zahlen werden Quantenzahlen auch durch Buchstaben beschrieben; es gelten die folgenden Symbole:
| n | l | |
|---|---|---|
| 0 | s | |
| 1 | K | p |
| 2 | L | d |
| 3 | M | f |
Unter einem „2p-Elektron“ ist danach ein Elektron der Hauptquantenzahl 2 und der Nebenquantenzahl 1 zu verstehen; „Elektronen der L-Schale“ sind Elektronen der Hauptquantenzahl 2. Bei der Besetzung der Energieniveaus gilt das Pauli-Verbot: Es stimmen niemals alle vier Quantenzahlen von Elektronen eines Atoms überein. Die Hauptquantenzahl 1 kann daher für höchstens zwei Elektronen zutreffen (n = 1; l = 0; ml = 0; ms = +½ oder −½) und die Hauptquantenzahl 2 für höchstens acht Elektronen (n = 2; l = 0, 1; ml = 0 für l = 0, ml = −1, 0, +1 für l = 1; ms = +½ oder −½). Allgemein können 2 n2 Elektronen dieselbe Hauptquantenzahl aufweisen („in der selben Schale sein“), während sich bei vorgegebener Haupt- und Nebenquantenzahl die folgenden maximalen Elektronenzahlen ergeben:
2 Elektronen im Zustand ns, 6 bei np, 10 bei nd und 14 bei nf.
Die Energien der einzelnen Niveaus hängen von n und l ab; ihre Abfolge ist:
1s, 2s, 2p, 3s, 3p, 4s, 3d, 4p, 5s, 4d, 5p, 6s, …
Beachten Sie, dass hinter der in vielen Lehrbüchern gezeigten Eselsbrücke, die diese Reihenfolge aus einem Schachbrettmuster ableitet, kein Naturgesetz steht.
In einem Atom im Grundzustand besetzen die Elektronen die erlaubten Energieniveaus mit der niedrigsten Energie, wobei die Hundsche Regel beachtet wird: Zustände gleicher Energie werden zunächst mit jeweils einem Elektron besetzt.
Im Periodensystem der Elemente sind die Elemente in der Folge ihrer Elektronenzahlen (= Kernladungszahlen = Ordnungszahlen) angeordnet. Eine eindeutige Zuordnung ergibt sich durch das Moseleysche Gesetz, auf das weiter unten eingegangen wird.
Das gewohnte Langperiodensystem entsteht, wenn Blöcke entsprechend den Nebenquantenzahlen gebildet werden, wenn also alle s-Elemente untereinander, alle p-Elemente untereinander, usw., angeordnet werden; Elemente, deren Elektronen höchster Energie die gleiche Hauptquantenzahl aufweisen, werden dabei in eine Zeile (Periode) geschrieben:
| n | s-Block | d-Block | p-Block | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | … | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
| 1 | 1H | 2He | |||||||||
| 2 | 3Li | 4Be | 5B | 6C | 7N | 8O | 9F | 10Ne | |||
| 3 | 11Na | 12Mg | 13Al | 14Si | 15P | 16S | 17Cl | 18Ar | |||
| 4 | 19K | 20Ca | 21Sc | … | 30Zn | 31Ga | 32Ge | 33As | 34Se | 35Br | 36Kr |
Auf diese Weise kommen Elemente mit gleicher Elektronenanordnung in der äußersten Schale untereinander zu stehen; diese gleiche Anordnung der Elektronen der unvollständig gefüllten Schale, der Valenzelelektronen bedingt ähnliche chemische Eigenschaften solcher Elemente einer Gruppe des Periodensystems (beachte eine Abweichung von der Regel: Helium, He, steht trotz seiner beiden Elektronen im 1s-Zustand nicht über Beryllium, Be, sondern in Übereinstimmung mit seinen chemischen Eigenschaften in der Gruppe der Edelgase).
Die Elemente des s-Blockes und des p-Blockes werden als Hauptgruppenelemente bezeichnet; bei diesen entspricht die Periodennummer der Hauptquantenzahl der Elektronen, die im Verlauf der Periode in ihr entsprechendes Niveau eingefüllt werden; die Gruppennummer entspricht nach alter Zählung der Zahl der Valenzelektronen, bei neuer Zählweise weist ein p-Block-Element der Gruppennummer i gerade i − 10 Valenzelektronen auf. Die Gruppennummer 18 entspricht einer vollständigen Auffüllung der s- und p-Niveaus einer Schale (Ausnahme: Für n = 1 ist die Schale mit zwei Elektronen gefüllt). Dies ist die Edelgaskonfiguration, die besonders stabile Elektronenanordnung der Edelgase.
Bei den Elementen des d-Blockes, den Nebengruppenelementen, werden Elektronen der Hauptquantenzahl n − 1 eingefüllt, obwohl schon Elektronen mit der Hauptquantenzahl n vorhanden sind. Die Elemente der ersten neun Nebengruppen, bei denen das d-Niveau nicht vollständig gefüllt ist, werden auch als Übergangselemente oder Übergangsmetalle bezeichnet. Bei den Elementen des f-Blockes schließlich werden Elektronen der Hauptquantenzahl n − 2 aufgefüllt.
Die 10 Elektronen des Edelgases Neon bilden die Elektronenkonfiguration 1s2 2s2 2p6. Die energetische Abfolge spiegelt die hohe Kernladungszahl des Neons im Vergleich zu Wasserstoff wider. Das Valenzelektronenkonzept wird deutlich, wenn die besondere Stabilität des 1s-Niveaus betrachtet wird (Abbildung auch als pdf):
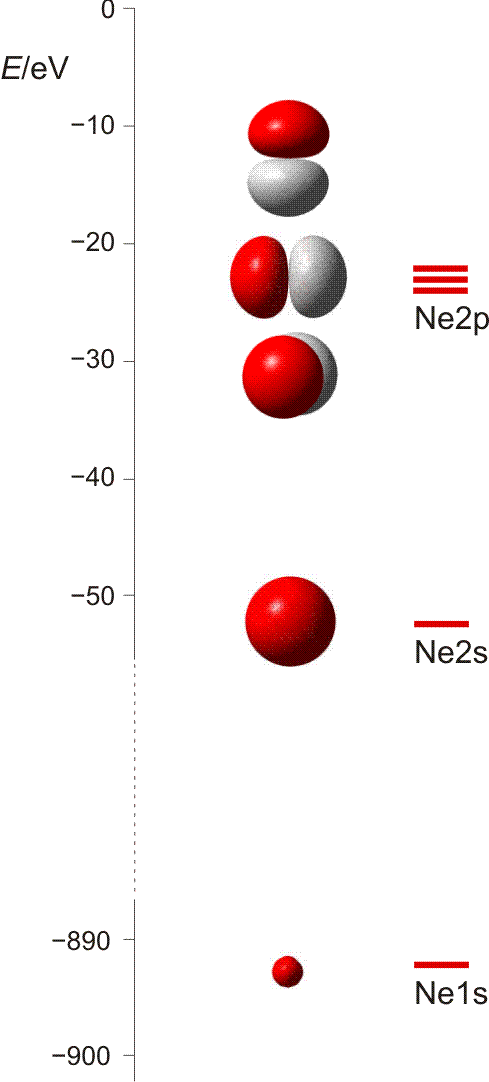
Die fünf besetzten Atomorbitale des Neons. Jedes Orbital ist mit einem spingekoppelten Elektronenpaar besetzt. Man beachte die starke Kontraktion und die hohe Stabilität des 1s-Orbitals.
In der Abbildung ist bei den einzelnen Orbitalen eine Gestalt angegeben. Im Teilchenmodell ist das Orbital mit dem Aufenthaltsraum des Elektrons verknüpft. Für die Deutung der chemischen Bindung im nächsten Kapitel ist es nützlicher, die durch n, l und ml charakterisierten Orbitale als dreidimensionale stehende Wellen anzusehen. Im Wellenmodell kommt zur Kontur des Orbitals eine Vorzeicheninformation hinzu, die bei einer zweidimensionalen Welle dem Wellenberg und -tal entspricht. In der Abbildung sind die verschiedenen Vorzeichen durch die Farben rot und grau unterschieden.
Versuch 6-3: Eindimensional stehende Wellen
Das 1s-Orbital ist kugelsymmetrisch, die zugehörige Wellenfunktion hat an jedem Ort des Raums ein positives Vorzeichen. Dasselbe scheint für das 2s-Orbital zu gelten, ein Schnitt durch das Orbital würde jedoch in Kernnähe einen Vorzeichenwechsel zeigen. Aufgrund der Kernnähe des Vorzeichenwechsels spielt dieser für die folgenden Bindungsbetrachtungen keine Rolle, da die Wechselwirkungen zwischen den Orbitalen zweier Atome durch die äußere Kontur dominiert wird.
Das 2p-Orbital zeigt einen Vorzeichenwechsel in der äußeren Kontur. Das Orbital besitzt eine durch den Kern verlaufende Knotenebene, in der die Wahrscheinlichkeit, das Elektron anzutreffen, Null ist. Man beachte, dass diese Aussage mit einer Teilchenvorstellung unverträglich ist. Die drei energiegleichen, „entarteten“ (engl. degenerate) p-Orbitale unterscheiden sich durch ihre Ausrichtung im Raum. Den drei Werten für ml entsprechen drei Orbitalausrichtungen entlang der drei Basisvektoren eines kartesischen Koordinatensystems. Zur Unterscheidung der drei p-Orbitale dienen alternativ zu ml = −1, 0 oder 1 die Bezeichnungen px, py und pz.
Man beachte eine im Folgenden wichtige Eigenschaft der gezeigten Neon-Orbitale: Werden zwei beliebige Orbitale einander überlagert, so ist der gemeinsame Raum, in dem gleiche Vorzeichen zusammentreffen und die Wellen einander verstärken (konstruktive Interferenz) genauso groß wie der Raum, in dem die Wellenfunktionen verschiedenes Vorzeichen haben und einander auslöschen (destruktive Interferenz) – die Nettoüberlappung ist 0, die Orbitale sind orthogonal.
Bei den folgenden Diskussionen der chemischen Eigenschaften von Atomen und Molekülen wird die Orbitalenergie im Bereich der Valenzelektronen eine wichtige Rolle spielen. Orbitalenergiediagramme verdeutlichen Trends im PSE, zum Beispiel bei der Ionisierungsenergie von Atomen oder deren Elektronenaffinität.
Die 1. Ionisierungsenergie („1. Ionisierungspotential“) I1 eines Atoms ist die Energie, die mindestens benötigt wird, um ein Elektron vollständig aus dem Atom zu entfernen. Aus dem Atom entsteht ein einfach positiv geladenes Ion:
X → X+ + e−
Die 1. Ionisierungsenergie entspricht weitgehend der Orbitalenergie des energiereichsten Elektrons. Die Entfernung des zweiten Elektrons erfordert die Energie I2, des dritten I3, und so fort. Ionisierungsenergien sind ein Maß für die Festigkeit, mit der Elektronen im Atom gebunden sind. I1 wächst innerhalb einer Periode von links nach rechts; das heißt, dass die Entfernung eines Elektrons umso schwerer fällt, desto kleiner ein Atom ist, desto näher also das betrachtete Elektron dem anziehenden Kern ist – siehe die vorige Abbildung. Für ein gegebenes Element wachsen die Ionisierungspotentiale in der Reihe I1, I2, I3, … schnell an, da die Entfernung von Elektronen aus einem zunehmend höher positiv aufgeladenen Atom immer mehr Energie erfordert. Die Entfernung eines Elektrons aus einer Edelgasschale bedingt einen auffälligen Sprung in der Folge der Ionisierungspotentiale (Abbildung auch als pdf).
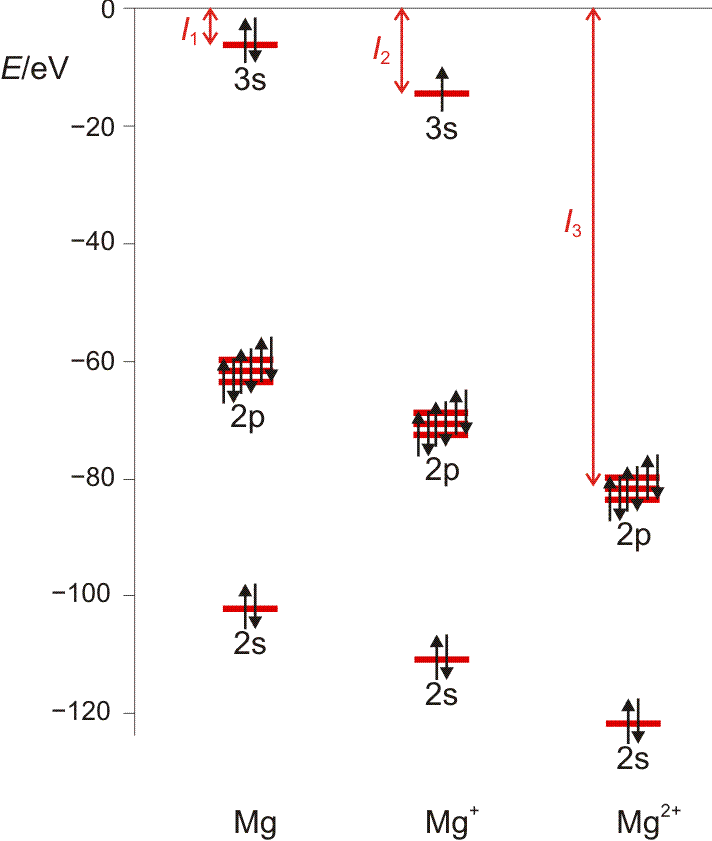
Orbitalenergien für Mg, Mg+ und Mg2+. Die Ionisierungsenergie wird benötigt, um das energiereichste Elektron zu entfernen. Man beachte den Sprung beim Wegfall der Valenzschale (technische Anmerkung: Orbitalenergien mit hf/6-31g(d,p), Mittelwerte für α- und β-Spin bei Mg+).
Die Elektronenaffinität (EA) eines Atoms ist die Energie, die frei wird oder benötigt wird, wenn ein Atom ein Elektron anlagert. Aus einem neutralen Atom wird dabei ein Anion:
X + e− → X−
Der enge Bezug zur Ionisierungsenergie wird klar, wenn der umgekehrte Vorgang betrachtet wird:
X− → X + e−
Die Elektronenaffinität erscheint nun vom Betrag her als Ionisierungsenergie des Anions; sie entspricht damit weitgehend der Orbitalenergie des höchsten besetzten Orbitals des Anions X−. Die folgende Abbildung (pdf) zeigt I1 und EA im Orbitalschema des Fluors:
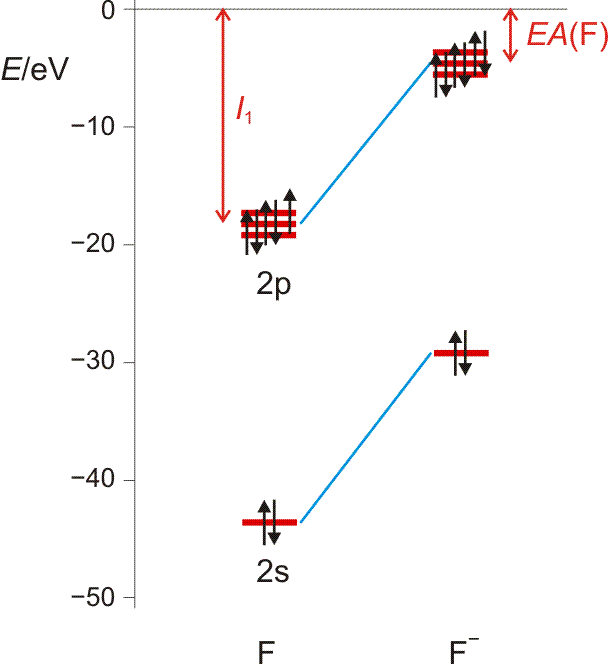
Die größten EA-Werte werden bei den Halogenen gefunden, die durch die Aufnahme eines Elektrons die Elektronenkonfiguration des im Periodensystems folgenden Edelgases erreichen. I1 und EA eines Atoms werden weiter unten noch einmal aufgegriffen, um bei der Behandlung der chemischen Bindung den wichtigen Begriff „Elektronegativität“ zu definieren.
Die konstruktive Interferenz einfach besetzter Atomorbitale führt zu bindenden Molekülorbitalen. Daneben sind nichtbindende und antibindende Kombinationen von Atomorbitalen zu beachten, wenn die Valenzelektronen zweier Atome in Wechselwirkung treten, um σ- und π-Bindungen aufzubauen. Die durch eine Lewis-Formel darstellbare kovalente 2e-2z-Bindung steht im Mittelpunkt der Valenzbindungsmethode („VB-Methode“), die aus der allgemeineren Molekülorbitalmethode („MO-Methode“) abgeleitet werden kann.
| Mortimer: 2, 6 Binnewies: 5 Riedel: 2.2 |
Wasserstoff kommt in Form von H2-Molekülen vor. Zwei H-Atome sind in diesen durch eine kovalente Bindung (Atombindung) miteinander verbunden. Die Ausbildung der Bindung lässt sich deuten, wenn wie bei der Besetzung von Atomorbitalen im vorigen Kapitel nach Molekülorbitalen gefragt wird, bei denen Elektronen nun aber mit mehr als einem Atomkern wechselwirken. Ein gedanklich naheliegendes Verfahren zur Konstruktion solcher Molekülorbitale (MOs) verbirgt sich hinter dem Kürzel LCAO (linear combination of atomic orbitals, Linearkombination von Atomorbitalen). Hierbei wird von Atomorbitalen ausgegangen und deren Wechselwirkung miteinander untersucht. Das Bild der stehenden Welle hilft dabei, eine Verstärkung oder eine Schwächung der Welle zu identifizieren (konstruktive oder destruktive Interferenz). Das Prinzip geht aus der folgenden Abbildung hervor.
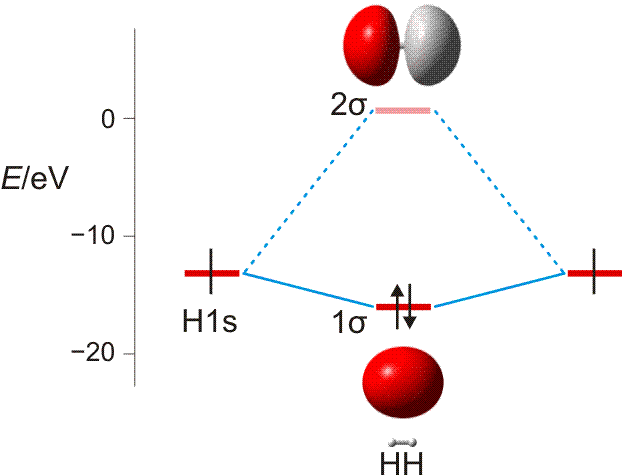
MO-Diagramm für H2. (Als Lage des antibindenden Orbitals ist die Energie des niedrigsten unbesetzten MOs des stabilen 1σ2 2σ0-Grundzustandes eingezeichnet.)
Technische Anmerkung: H2-Energien mit hf/aug-cc-pv5z bei 73,37 pm Kernabstand.
In zwei räumlich getrennten H-Atomen liegt das jeweils eine Elektron im kugelsymmetrischen 1s-Zustand vor. Nähern sich die beiden Atome nun einander, so gibt es zwei Möglichkeiten der Wechselwirkung: die Wellen verstärken einander (gleiches Vorzeichen der Wellenfunktion) oder sie löschen einander aus (verschiedenes Vorzeichen). Die erste Möglichkeit führt zu einer Lokalisierung der Elektronen zwischen den positiv geladenen Kernen und damit zur Bindung. Elektronen, die sich in diesem MO befinden, sind aufgrund ihrer Wechselwirkung mit zwei statt einem Kern gegenüber dem freien Atom stabilisiert, sie liegen auf der Energieachse unterhalb der Atomorbitalenergie – das MO ist bindend. Das Gegenteil trifft zu, wenn die beiden Wellen mit verschiedenem Vorzeichen miteinander kombiniert werden: es entsteht ein MO, das auf der Kernverbindungslinie eine Knotenebene aufweist. Ist dieses antibindende Orbital mit Elektronen besetzt, kommt es zur Lokalisierung von Elektronen jenseits der Proton–Proton-Achse, was einem unstabilen Zustand entspricht (man betrachte den Zerfall von angeregten H2-Molekülen in der oben behandelten Plücker-Röhre). Man beachte eine wichtige Regel: die Gesamtzahl der im Schema berücksichtigten Atomorbitale (hier 2) entspricht der Zahl der gebildeten Molekülorbitale (hier mit 1σ und 2σ auch 2).
Der letzte Schritt bei der Konstruktion eines MO-Schemas ist das „Einfüllen“ der vorhandenen Elektronen. Analog dem Aufbauprinzip werden diese unter Beachtung von Pauli-Prinzip und Hundscher Regel auf möglichst stabile Zustände verteilt. Beim H2-Molekül wird dabei das bindende MO mit einem spingekoppelten Elektronenpaar gefüllt während das antibindende MO leer bleibt – es ist eine kovalente Bindung (Elektronenpaarbindung, Atombindung) entstanden. Das bindende MO ist dabei rotationssymmetrisch, es liegt eine σ-Bindung vor (vergleichen Sie die Symmetrieeigenschaften eines s-Orbitals). Die beiden Orbitale des Schemas werden mit 1σ und 2σ bezeichnet, üblich ist auch σ und σ* (sprich „sigma-Stern“), wobei der * das Orbital als antibindend markiert. Die Notation der Elektronenkonfiguration entspricht völlig dem Gebrauch bei Atomen; für den Grundzustand des H2-Moleküls 1σ2 2σ0.
Ein weiterer wichtiger Begriff ist die Bindungsordnung. Hierzu addiert man die Zahl der Elektronen in bindenden Molekülorbitalen (solche ohne Knotenebene senkrecht zur Kern-Kern-Achse), subtrahiert dann die Zahl der Elektronen in antibindenden MOs (solche mit Knotenebene senkrecht zur Kern-Kern-Achse) und dividiert durch zwei. Bei H2:
BO = (2 − 0)/2 = 1
Die beiden Wasserstoffatome werden durch eine Einfachbindung zusammengehalten.
Im folgenden Kapitel werden wir die Reaktion von Wasserstoff mit Chlor, Cl2, untersuchen. Die beiden Gase reagieren exotherm zu ebenfalls gasförmigem Chlorwasserstoff (Hydrogenchlorid), HCl, mit dessen Hilfe das Prinzip der nichtbindenden Wechselwirkung verdeutlicht werden soll.
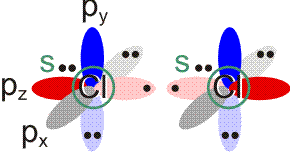
Im Gegensatz zum Wasserstoffatom sind beim Chloratom aufgrund der Elektronenkonfiguration 1s2 2s2 2p6 3s2 3p5 auch p-Orbitale zu beachten. Gegenüber dem H2-Fall treten nun zwei neue Aspekte auf: (1) Welche Wechselwirkung tritt zwischen dem H(1s)- und den Cl(3p)-Orbitalen auf? (2) Wie ist die Wechselwirkung zwischen dem H(1s)- und dem Cl(3s)-Orbital, die auf der Energieachse recht weit auseinander liegen. Die folgende Abbildung zeigt die Antwort:
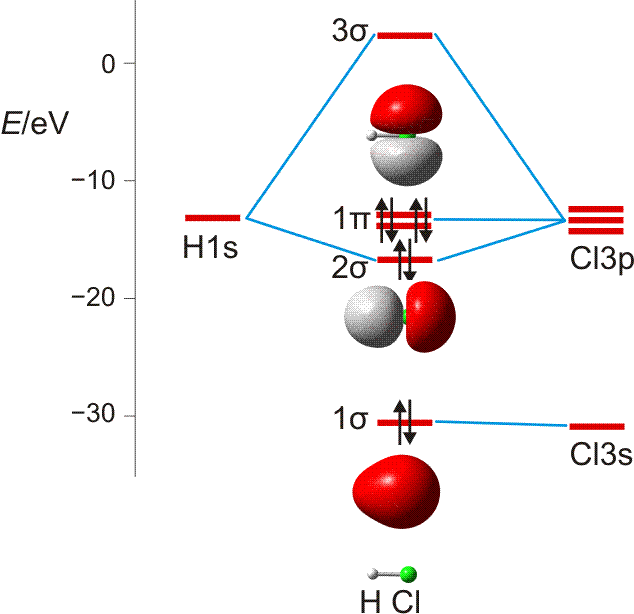
MO-Schema für HCl, Elektronen in Atomorbitalen weggelassen (technische Anmerkung: rmp2/6-31g(d,p), bei Cl über α- und β-Spins gemittelt, Ladung auf Cl nach Mulliken-Populationsanalyse: −0.17 e)
Von den drei 3p-Orbitalen des Chlors kombiniert nur eines mit dem H(1s)-Orbital zu einem bindenden (2σ) und einem antibindenden (3σ) MO. Es ist gerade dasjenige, das rotationssymmetrisch zur H-Cl-Bindungsachse liegt (die Wechselwirkung ist „symmetrieerlaubt“). Wird die H-Cl-Bindungsachse in einem kartesischen Koordinationsystem als z-Achse gewählt, so handelt es sich um das 3pz-Orbital.
Wird nun die Wechselwirkung zwischen dem H(1s)-Orbital und dem 3px- oder 3py-Orbital des Chlors betrachtet, so wird jeder konstruktiv interferierende Bereich durch einen gleich großen destruktiv interferierenden Bereich kompensiert. Die Orbitale sind orthogonal, die Wechselwirkung ist nichtbindend oder „symmetrieverboten“. Die beiden nichtbindenden p-Orbitale erscheinen im Schema energetisch unverändert (was nicht zwingend ist, vgl. weiter unten bei HF) und ohne Wechselwirkung mit dem H(1s)-Orbital.
Vom Ergebnis her ähnlich ist die H(1s)-Cl(3s)-Wechselwirkung im Diagramm behandelt. Das 1σ-Orbital erscheint bei fast derselben Energie wie das Cl(3s)-Orbital, außerdem scheint das H(1s)-Orbital nur wenig zur äußeren Gestalt von 1σ beizutragen – das ursprünglich kugelsymmetrische Cl(3s)-Orbital ist im 1σ-Orbital noch deutlich zu erkennen, allerdings ist es in Richtung auf das H-Atom „ausgebeult“. Das Orbital lässt sich in sehr grober Näherung als nichtbindend einstufen und dem Chloratom zuordnen. Es handelt sich dabei um eine quantitative Feststellung, denn prinzipiell führt die symmetrieerlaubte Wechselwirkung zu einer bindend-antibindend-Kombination und nicht zu einer streng nichtbindenden, symmetrieverbotenen Wechselwirkung – wir werden gleich darauf zurückkommen.
Zuerst aber das Ergebnis in der Übersicht: Je größer die Energiedifferenz zwischen zwei Orbitalen ist, desto kleiner ist die energetische Aufspaltung der MOs. Das Elektronenpaar ist räumlich an dem Atom lokalisiert, welches das Atomorbital mit fast gleicher Energie beisteuert. Bei der Berechnung der Bindungsordnung werden nichtbindende Orbitale nicht mitgezählt. Fünf Atomorbitale (1 von H, 4 von Cl) ergeben 1 bindendes, 1 ziemlich nichtbindendes, 2 streng nichtbindende und 1 antibindendes, insgesamt also 5 MOs, deren stabilere 4 jeweils 2 Elektronen enthalten. Es gilt:
BO = {2 (in 2σ) − 0 (in 3σ)}/2 = 1
Eine quantitative Aussage zur H(1s)-Cl(3s)-Wechselwirkung ist im MO-Formalismus wenig anschaulich. Im übernächsten Abschnitt wird daher zuerst am Beispiel des Cl2-Moleküls eine Methode vorgestellt, welche die MO-Theorie mit der nun zuerst vorgestellten Valenzbindungsmethode verbindet.
Lewis-Formeln erlauben die knappe Fornulierung von Elektronenpaarbindungen. Dargestellt werden die Elektronen der Valenzschale, ein Strich symbolisiert ein spingekoppeltes Elektronenpaar, ein Punkt ein ungepaartes Elektron. Die Elektronenkonfigurationen der Atome H (1s1), N (2s2 2p3 = 2s2 2px1 2py1 2pz1), O (2s2 2p4 = 2s2 2px2 2py1 2pz1), F (2s2 2p5 = 2s2 2px2 2py2 2pz1) und Cl (3s2 3p5 = 3s2 3px2 3py2 3pz1) im Grundzustand wird dargestellt als:
Bindungen entstehen durch Spinkopplung ungepaarter Elektronen zu Elektronenpaaren:
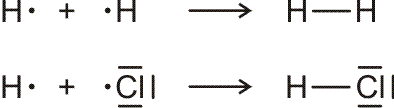
Lewis-Formeln sind oft hinreichend geeignet, das Ergebnis der Valenzbindungsmethode (engl. valence bond method, VB-Methode) darzustellen. Während bei der Molekülorbitalmethode sich über das ganze Molekül erstreckende Molekülorbitale (meist nach der LCAO-Methode) ermittelt werden, um anschließend die vorhandenen Elektronen nach dem Aufbau-Prinzip einzufüllen, sind Elektronenpaare bei der VB-Methode an einem Atom (einsame Elektronenpaare) oder zwischen zwei Atomen (bindende Elektronenpaare) lokalisiert. (Eine Delokalisierung über mehrere Atome kann oft durch „mesomere Grenzstrukturen“ beschrieben werden, auf die weiter unten eingegangen wird.)
Bei H2 und HCl führt das in den Orbitaldiagrammen zusammengefasste Verfahren, das im Wesentlichen der MO-Methode entspricht, aufgrund des nur einen Valenzorbitals an jedem H-Atom zu einem Ergebnis, das sehr dicht bei einer VB-Behandlung liegt. So ist es naheliegend, das 1σ-Orbital des H2-Moleküls als das bindenden Elektronenpaar der Lewis-Formel zu identifizieren. Beim HCl-Molekül zeigt das MO-Schema mit drei chlorständigen Orbitalen (1 π und 1σ) eine sehr ähnliche Situation wie die Lewis-Formel mit 3 einsamen Elektronenpaaren (engl. lone pairs). Das bindende Elektronenpaar der Lewis-Formel findet sich im MO-Schema im Wesentlichen als 2σ wieder.
Um den engeren Bezug von MO- und VB-Methode darzustellen, betrachten wir nun das Chlor-Molekül.
Am Chlormolekül, Cl2, soll gezeigt werden, dass es einen Zusammenhang zwischen MO- und VB-Behandlung gibt. Hierzu zuerst die VB-Methode. Ausgangspunkt sind zwei Chloratome und deren 3s23p5-Valenzkonfiguration. Nach der VB-Methode ist eine σ-Einfachbindung durch die Wechselwirkung der einfach besetzten pz-Orbitale zu erwarten:
Als Lewisformel ausgedrückt:
Auch die MO-Methode geht von den Atomorbitalen aus und kombiniert diese zu bindenden und antibindenden MOs:
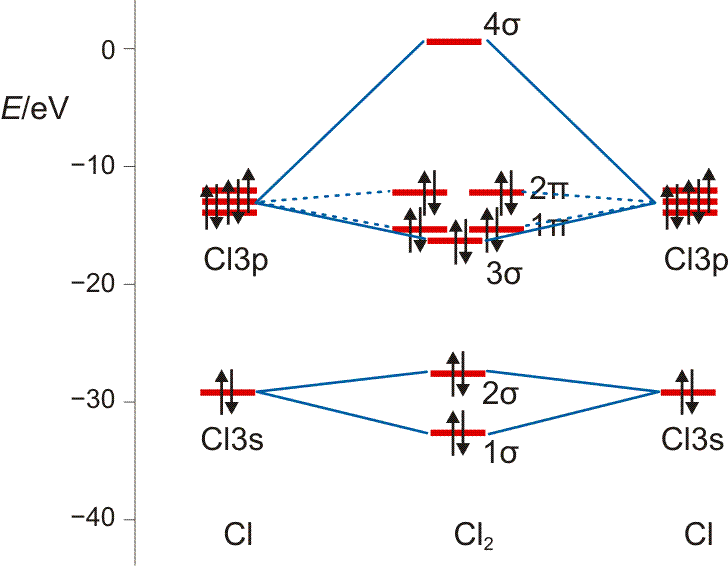
Wie bei H2 und HCl treten σ-Wechselwirkungen auf – sowohl bindende durch 3s-3s- und 3pz-3pz-Überlappung als auch eine antibindende 3s-3s-Wechselwirkung. Zusätzlich kommt durch seitliche Überlappung vom Typ 3px-3px und 3py-3py eine weitere Wechselwirkung hinzu, nämlich bindende und antibindende π-Molekülorbitale, die im Gegensatz zu den σ-Orbitalen nicht rotationssymmetrisch um die Bindungsachse sind, sondern in der Bindungsachse eine Knotenebene aufweisen.
Im folgenden Schema sind auf der linken Seite alle Molekülorbitale des Energiediagramms abgebildet. Zusätzlich ist das Ergebnis eines Rechenverfahrens gezeigt, mit dem immer dann eine VB-typische Betrachtung aus einer MO-Analyse abgeleitet werden kann, wenn sich die Valenzelektronen des betreffenden Moleküls in normale kovalente Bindungen und freie Elektronenpaare aufteilen lassen. Hierzu werden MOs (die zuvor durch Linearkombination aus AOs errechnet wurden) untereinander linearkombiniert. Für das Cl2-Molekül ist das Ergebnis besonders übersichtlich:
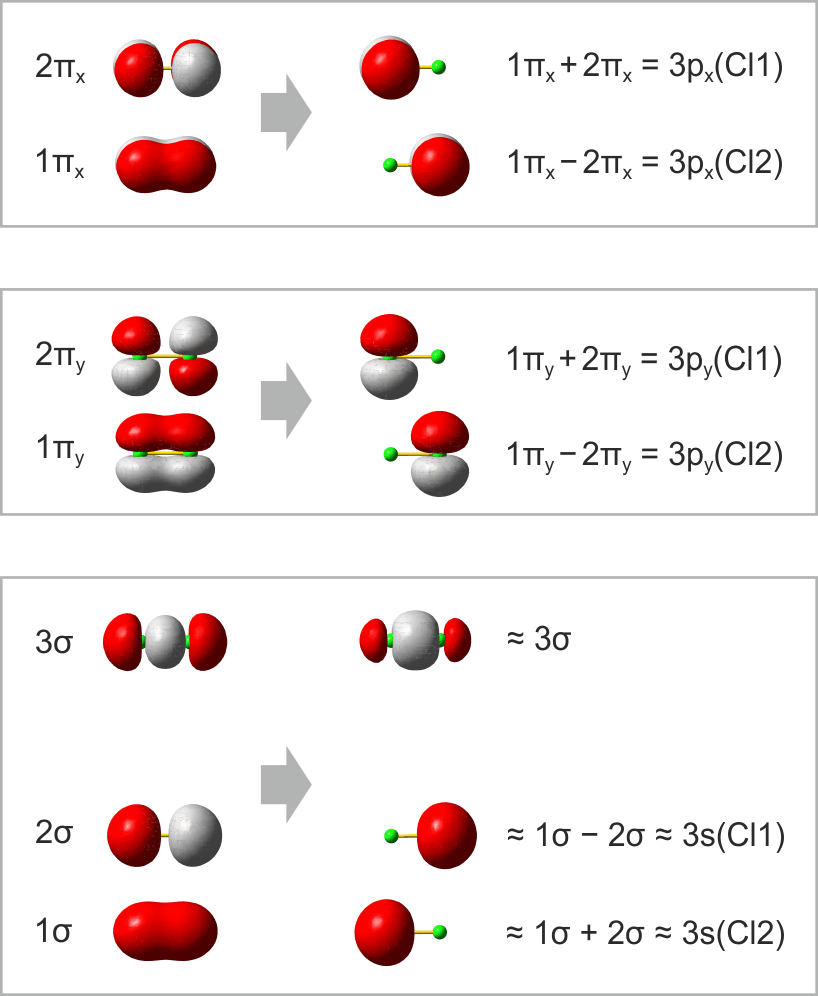
Das Ergebnis dieser Lokalisierung von MOs erschließt sich beim Vergleich mit der Lewis-Formel unmittelbar: ist in einem Molekül sowohl ein bindendes MO als auch das dazugehörige antibindende MO besetzt (linke Seite des Diagramms), so führt die Linearkombination des Paares zu jeweils einem freien Elektronenpaar an jedem Atom (rechte Diagrammseite). Da diese Bedingung für alle Orbitale außer dem 3σ-MO zutrifft, bleibt dieses als einziges beiden Chlor-Atomen zugeordnet. In der Lewis-Formel entspräche es dem bindenden Elektronenpaar, während die übrigen sechs Elektronenpaare mit den freien Elektronenpaaren der Lewisformel gleichgesetzt werden können.
Ein besonders nützliches Ergebnis des Verfahrens besteht in einer quantitativen Aussage, in welchem Umfang die Bindung – die vor allem durch die 3pz-3pz-Wechselwirkung entsteht – Anteile des Typs 3s-3pz enthält. (Vollziehen Sie als Vorübung nach, dass das auf der rechten Seite mit ≈3σ bezeichnete Orbital aus dem 3σ-MO entsteht, wenn in geringem Umfang 1σ subtrahiert wird). Für Cl2 bestätigt das Ergebnis den Eindruck, dass die Bindung vor allem 3pz-3pz-Charakter hat, indem jedes 3s-Atomorbital zu weniger als 10 % zur Bindung beiträgt.
Für das HCl-Molekül ist der Anteil höher. Die im MO-Schema von HCl sichtbare „Beule“ im 1σ-Orbital entspricht einem Anteil der H(1s)-Cl(3s)-Wechselwirkung von ca. 15 %, die wir näherungsweise vernachlässigt hatten. Der gemeinsame Beitrag von s- und p-Orbitalen zu einer Bindung wird im VB-Modell unter dem Stichwort „Hybridisierung“ behandelt. Wir führen dieses Konzept am Beispiel des N2-Moleküls ein, da dort die s-p-Wechselwirkung deutlich stärker ist.
Stickstoff ist der reaktionsträge Hauptbestandteil der Luft. Wie Wasserstoff, Chlor und auch Sauerstoff besteht Stickstoff aus zweiatomigen Molekülen. Die Elektronenkonfiguration des N-Atoms ist 1s2 2s2 2p3. Bei einer VB-Behandlung würden die drei ungepaarten p-Elektronen eines jeden Stickstoffatoms zu Elektronenpaarbindungen spingekoppelt:
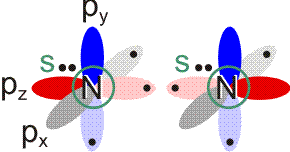
Durch Spinkopplung der beiden pz-Elektronen entsteht wie beim Cl2-Molekül eine σ-Bindung. Die beiden verbleibenden ungepaarten Elektronen eines jeden N-Atoms lassen sich zu zwei π-Bindungen koppeln. Insgesamt resultiert die Bindungsordnung 3. Das spingekoppelte Elektronenpaar im Grundzustand des N-Atoms (2s2) erscheint im Molekül als einsames Elektronenpaar bei jedem der beiden N-Atome. Mit Lewis-Formeln dargestellt:
Auch bei diesem nur zweiatomigen Teilchen liegt die MO-Behandlung noch nahe bei der VB-Betrachtung: Bei der Suche nach symmetrieerlaubten Wechselwirkungen führen die gleichen Überlegungen wie bei Cl2 zu zwei Gruppen von Atomorbitalen (die Bindungsachse sei auch hier die z-Achse): insgesamt vier 2s- und 2pz-Orbitale können in symmetrieerlaubter Weise kombiniert werden und ebenfalls vier 2px- und 2py-Orbitale liegen in nichtbindender Wechselwirkungen zu den Orbitalen der ersten Gruppe vor. Untereinander können die beiden 2px- und die beiden 2py-Orbitale zu zwei π-Wechselwirkungen kombiniert werden: zu bindenden MOs mt einer Knotenebene in der Molekülachse (1π) und antibindenden MOs mit einer Knotenebene in der Molekülachse und einer Knotenebene senkrecht zur Kernverbindungslinie (2π). In der Abbildung ist nur die π-Wechselwirkung in der Zeichenebene gezeigt (πy mit dem Koordinatensystem der VB-Betrachtung), nicht aber das dazu senkrecht stehende πx-Orbital.
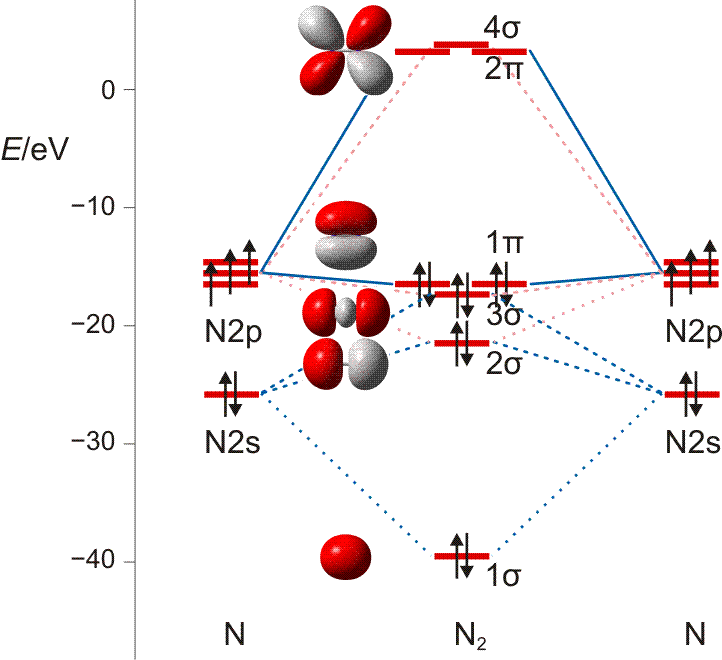
MO-Diagramm für N2 ( pdf). Es ist jeweils nur ein 1π und 2π-Orbital eingezeichnet. (Technische Anmerkung: das 2s-Orbital ist beim Mittelwert von α- und β-Spin eingezeichnet.)
Als Bindungsordnung ergibt sich durch Summieren über die aus 2 × 4 Atomorbitalen gebildeten 8 MOs:
BO = {2 (1σ) − 2 (2σ) + 2 (3σ) + 4 (1π) − 0 (2π) − 0 (4σ)}/2 = 3
Im Gegensatz zu Cl2 zeigen die σ-Wechselwirkungen nun eine merkliche Durchmischung von 2s- und 2p-Anteilen. Diese entdecken Sie auch dann, wenn Sie zuerst nur das MO-Schema analysieren.
Bei der VB-Methode wird die s-p-Wechselwirkung als Eigenschaft der Atomorbitale betrachtet. In der Praxis werden die MOs hierzu lokalisiert. Die beiden π-Bindungen bleiben hierbei als solche erhalten, da im Gegensatz zu Cl2 keine besetzten antibindenden π-Orbitale zur Linearkombination der MOs zur Verfügung stehen. Bei den σ-Orbitalen ist zu erwarten, dass das 1σ-2σ-Paar zu zwei freien Elektronenpaaren kombiniert werden kann. Die Rechnung ergibt das folgende Bild (links die MOs, rechts die lokalisierten Orbitale; unter beiden Orbitalgruppen ist zur Orientierung ein N2-Rumpf eingezeichnet):
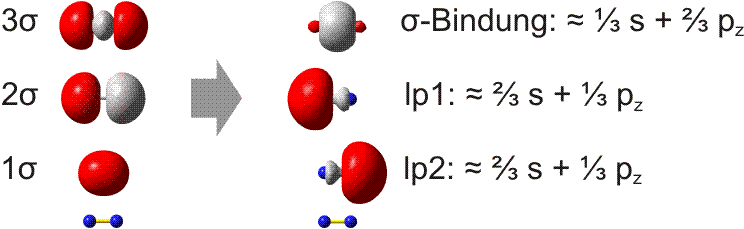
Weder das σ-Bindungsorbital noch die beiden freien Elektronenpaare (im Bild: lp1 und lp2) haben die Form, die für eine Wechselwirkung zwischen reinen s- oder p-Orbitalen zu erwarten ist. Die Rechnung ergibt dementsprechend die in der Abbildung angegebenen s- und p-Anteile. In der VB-Methode wird nun ein anschaulicher Formalismus eingeführt: die s-p-Mischung wird vor der Linearkombination zu Bindungen vorgenommen, indem s- und p-Atomorbitale zu Hybridorbitalen linearkombiniert werden. So entsteht eine Bindung mit zum Beispiel 35% s- und 65% p-Charakter durch Wechselwirkung zweier Hybridorbitale mit genau diesen s- und p-Anteilen:
Man beachte, dass die Summe der s- und p-Anteile der Gesamtzahl der Orbitale entsprechen muss: beim N2-Molekül addieren sich daher 1/3 s-Anteil der σ-Bindung und 2/3 s-Anteil des freien Elektronenpaars zu 1. Dasselbe gilt für das pz-Orbital. Das konkrete Ausmaß der s-p-Mischung ist eine charakteristische Variable in der Chemie der Hauptgruppenelemente. In Teil B der Vorlesung werden Trends im Periodensystem behandelt.
Die Beschreibung der chemischen Bindung durch den Lewis-Formalismus ist das einfachste Hilfsmittel, um ein Molekül darzustellen. Wenn sich lediglich die Frage nach Basisinformationen zu Molekülbau und -eigenschaften stellt (Ist das Molekül gewinkelt oder linear? Hat es ein Dipolmoment oder nicht?), reichen Lewis-Formeln oft aus.
Als Mindestanforderung an die Darstellbarkeit eines Moleküls durch eine Lewis-Formel sollte dabei verlangt werden, dass – notfalls durch die Formulierung der später im Detail behandelten „mesomeren Grenzstrukturen“ – (1) die Bindungsordnung und (2) der Spinzustand (Zahl der ungepaarten Elektronen) korrekt wiedergegeben wird. Von den besprochenen Molekülen trifft dies auf H2, HCl, Cl2 und N2 zu. Schon bei dem für das Leben wichtigsten Luftbestandteil, dem Sauerstoff, kommt die Lewis-Formulierung zu einer falschen Aussage.
Versuch 9-1: Paramagnetismus von Sauerstoff • ein mit flüssigem Sauerstoff gefülltes Reagenzglas wird deutlich in das Feld eines Elektromagneten hineingezogen) und Diamagnetismus von Stickstoff (ein mit flüssigem Stickstoff gefülltes Reagenzglas scheint unter diesen Bedingungen nicht auf das Magnetfeld zu reagieren.
O2 ist im Grundzustand paramagnetisch. Diese Eigenschaft zeigt, dass im O2-Molekül ungepaarte Elektronen vorhanden sind. Eine quantitative Behandlung zeigt, dass zwei ungepaarte Spins pro Molekül vorliegen. Dies ist nach der einfachen Formulierung einer Lewisformel nicht zu erwarten:
Allerdings zeigt sich schon bei einer näheren Betrachtung der zugrunde liegenden Atomorbitalwechselwirkungen, dass mit Schwierigkeiten gerechnet werden darf. Während sich nämlich bei N2 zwanglos zwei gleichwertige π-Bindungen aus vier gleichwertigen p-Orbitalen ergeben, zeigt dieselbe Ansicht für O2 ein Problem:
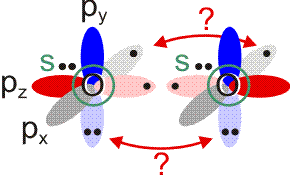
Obwohl geometrisch gleichwertig, sind die px- und py-Orbitale jetzt unterschiedlich besetzt – eines mit einem einzelnen Elektron, das andere mit einem Elektronenpaar.
Die MO-Rechnung ergibt ein Diagramm, das demjenigen des Stickstoffmoleküls sehr ähnlich ist. Wegen der um zwei größeren Valenzelektronenzahl ist nun jedoch der bei N2 leere 2π-Zustand mit zwei Elektronen besetzt und zwar wegen der Hundschen Regel mit jeweils 1 Elektron – O2 ist paramagnetisch. Die Beschreibung des Spinzustands ist als „Multiplizität“ gebräuchlich. Hierzu werden die ms-Werte zu einem Gesamtspin S addiert, anschließend wird die Multiplizität als 2S + 1 gebildet (für das Sauerstoffmolekül mit S = 2 × ½ = 1: 2 × 1 + 1 = 3). Teilchen mit einer Multiplizität von 1, 2, 3 sind im Singulett-, Dublett-, Triplett-Zustand. Die stabile Form von O2 ist also der Triplett-Sauerstoff.
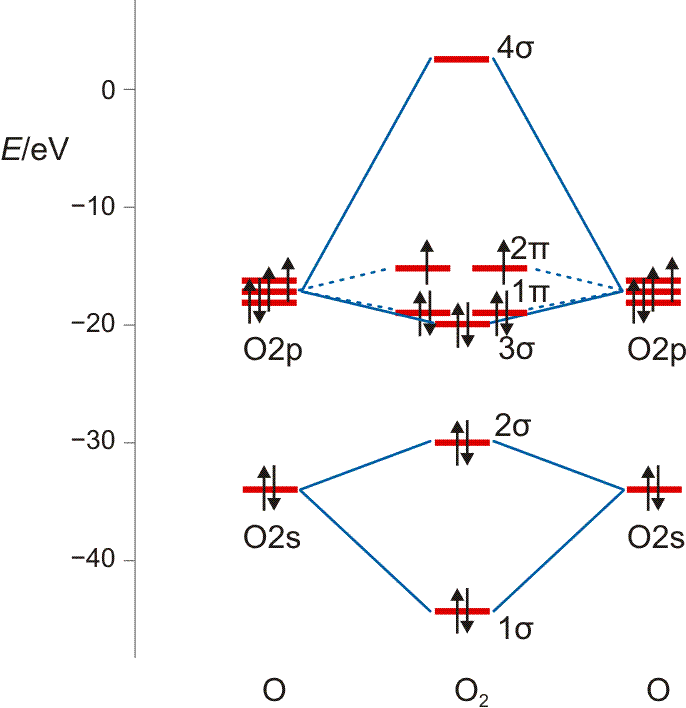
MO-Diagramm für O2.
Technische Anmerkung: O2-Energien und Atomabstand mit ump2/aug-cc-pvqz, 121,9 pm Atomabstand.
Eine Analyse der Bindungsordnung ergibt eine Doppelbindung:
BO = {2 (1σ) − 2 (2σ) + 2 (3σ) + 4 (1π) − 2 (2π)}/2 = 2
Sauerstoff, ein „Diradikal“, gehört zu den wenigen Molekülen, die stabil sind, ohne dass sich im Grundzustand für die Summe der (magnetischen) Spinquantenzahlen 0 ergibt, ohne also dass alle Elektronen spingepaart vorliegen. Zahlreiche chemische Reaktionen laufen nur ungehemmt ab (meist solche, bei denen keine Übergangsmetalle anwesend sind), wenn die Summe der ungepaarten Spins gleich bleibt. Entsteht bei einer solchen Reaktion Sauerstoff, so wird dieser nicht im Triplett-Grundzustand frei, sondern als unstabiler und äußerst reaktiver Singulett-Sauerstoff (den man mit der gezeigten Lewis-Formel beschreiben könnte).
Versuch 9.2: Singulettsauerstoff • Bei der Reaktion
H2O2 + Cl2 + 2 OH− → 1O2 + 2 Cl− + 2 H2O
wird Singulett-Sauerstoff gebildet. Bei der Rückkehr in den 3O2-Grundzustand wird rotes Licht der Wellenlängen 633 und 760 nm emittiert (190 = 2 × 95 und 158 kJ mol−1 entsprechend einer Emission zweier Singulett-Sauerstoff-Formen), hierzu Holleman-Wiberg: „… ist die Umsetzung von Hypochlorit mit Wasserstoffperoxid von einer Emission begleitet, die man mit dunkeladaptiertem Auge als roten Schimmer wahrnehmen kann.“
Das MO-Modell zeigte beim O2-Molekül seine Leistungsfähigkeit. Trotzdem kommt die Grundidee dieser Betrachtung – Orbitale über das gesamte Molekül zu delokalisieren – bei der Behandlung zweiatomiger Moleküle naturgemäß kaum zum Vorschein, da sich über nur zwei Atome nicht viel delokalisieren lässt. Viel klarer zeigen sich die Unterschiede zwischen MO- und VB-Methode, wenn mehratomige Moleküle betrachtet werden. Ein Musterbeispiel ist Methan, CH4.
Methan ist tetraedrisch aufgebaut, das heißt, die H-Atome bilden die Ecken eines Tetraeders, in dessen Schwerpunkt sich das Kohlenstoffatom befindet:
Schematisch (Erläuterung zur Schreibweise):
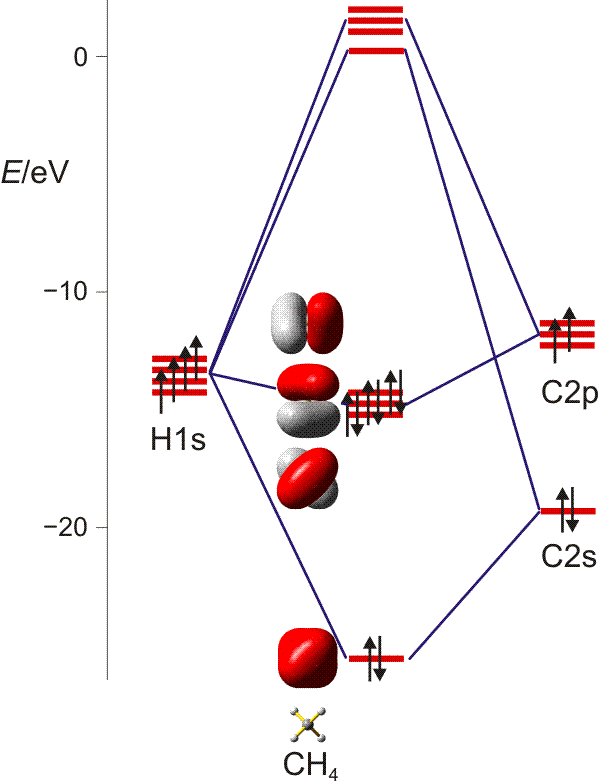
Bei der MO-Methode werden die Atomorbitale der vier H-Atome und des C-Atoms zu 5z-2e-Bindungen kombiniert – jedes Molekülorbital erstreckt sich also über alle Atome. Ausgehend von den Atomorbitalen des Kohlenstoffs wird hierzu jedes der vier Kohlenstoff-Valenzorbitale so mit einem Satz von jeweils vier H-ständigen Orbitalkombinationen linearkombiniert, dass sich vier bindende MOs ergeben. Bei den nicht besetzten antibindenden MOs sind die Vorzeichen der Wellenfunktionen vertauscht (entweder bei C oder beim H4-Teil):
Schematisch:
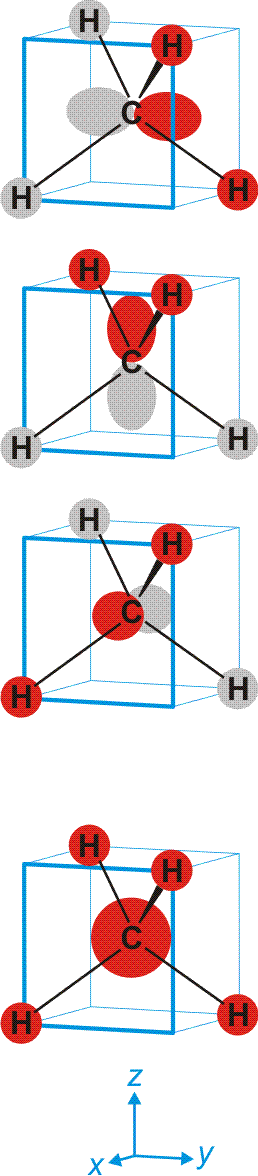
Man beachte, dass sich jeder einzelne der vier gleichwertigen C–H-Kontake aus vier Komponenten zusammensetzt. Da jedes bindende MO alle vier C–H-Kontakte einschließt, ist die Bindungsordnung für die einzelne C–H-Bindung 4 × ¼ = 1.
Wegen der hohen Symmetrie ist die Lokalisierung in 2z-2e-Bindungen übersichtlich: man überlagere zur Übung alle vier MOs des abgebildeten Schemas mit positivem Vorzeichen. Heraus kommt eine 2e-2z-Bindung, zu der nur das 1s-Orbital des H-Atoms vorne rechts oben beiträgt sowie ein Hybridorbital am Kohlenstoffatom, das ¼ s- und ¾ p-Charakter hat. Dies gilt für alle Bindungen in diesem hochsymmetrischen Molekül. Wegen des dreifach höheren Gewichts der p-Orbitale an den vier gleichartigen Bindungen werden diese „sp3-Hybridorbitale“ genannt. (Es ist eine lehrreiche Übung, sich graphisch klar zu machen, dass zwei senkrecht aufeinander stehende p-Orbitale durch eine 25-%-ige s-Beimischung ihre Orthogonalität verlieren. Diese wird wiederhergestellt, wenn durch Vergrößerung des Winkels zwischen den Orbitalen deren gegenseitige Überlappung im verstärkten Bereich vermindert und im geschwächten Bereich vergrößert wird. Beim „Tetraederwinkel“ von 109.5° ist wieder Orthogonalität erreicht.)
Werden, wie in Lehrbüchern üblich, die VB-Orbitale nicht durch Lokalisierung aus MOs abgeleitet, beginnt das Aufstellen der Formel mit einem Problem. Ein Kohlenstoffatom im Grundzustand scheint mit seiner Valenzelektronenkonfiguration 2s2 2p2 nicht über vier einfach besetzte Orbitale zu verfügen. Im ersten Schritt wird daher ein energiereicherer 2s1 2p3-Zustand formuliert. Die benötigte Zahl ungepaarter Elektronen steht nun zur Verfügung. Durch Linearkombination wird nun die 2s1 2p3Konfiguration zu einer (sp3)4-Konfiguration hybridisiert. Die vier gleichwertigen Hybridorbitale weisen nun in die Ecken eines Tetraeders.
Alle vier Bindungen im Methan-Molekül sind äquivalent. Im Sinne des VB-Modells sind alle vier Elektronenpaare gleich hybridisiert. Eine grundlegend einfachere Betrachtungsweise als VB- und MO-Methode ergibt sich, wenn diese Eigenart des Methans auf alle denkbaren Moleküle übertragen wird – wenn wir also das Cl2-Molekül so verstehen, dass das bindende Elektronenpaar ebenso wie die freien Elektronenpaare sp3-Hybridorbitale darstellen. Da wir nur wenige Moleküle kennenlernen werden, bei denen diese Vereinfachung zulässig ist, werden wir diesem VSEPR-Modell (valence shell electron pair repulsion, Valenzelektronenpaarabstoßung) keine allzu große Aufmerksamkeit schenken (auf den schwerer wiegenden Nachteil, die wichtige Oktettregel nicht zu beachten, kommen wir noch zu sprechen). Beispiele für Moleküle, bei denen dieses Modell erfolgreich angewendet werden kann, sind NH3 und H2O. Das VSEPR-Modell wird daher im Kapitel „Polare Moleküle“ vorgestellt.
In der Vorlesung werden wir das Bindungsmodell verwenden, dass hinreichend – aber nicht unnötig – komplex ist, um die gerade interessierende Frage angemessen zu beantworten. Oft wird zur Beschreibung eines Moleküls die Lewis-Formel ausreichen. Die MO-Methode bietet sich gerade für Mehrzentrenbindungen an, hier wird jedoch auch auf mesomere Grenzformeln eingegangen, schon deswegen, da diese in den Lehrbüchern weit verbreitet sind.
Das Beipiel Methan zeigt, in welchem Fall sich die MO-Methode besonders empfiehlt, nämlich wenn Aussagen zu Orbitalenergien unmittelbar abgeleitet werden sollen (vor einer Lokalisierung, mit Hilfe der MOs so wie sie sind – man nennt sie zur Unterscheidung von lokalisierten MOs „kanonische“ MOs). Wird zum Beispiel Methan mit ähnlichen spektroskopischen Methoden untersucht wie sie oben für das H-Atom besprochen wurden, so zeigen sich im Grundzustand besetzte Orbitale bei zwei Orbitalenergien und nicht – wie es eine oberflächliche VB-Betrachtung mit vier äquivalenten Hybridorbitalen suggeriert – nur einer einzigen (wobei eine sorgfältige VB-Analyse bei dieser äquivalenten Methode natürlich auch zum richtigen Ergebnis führt, aber eben nicht auf den ersten Blick). Wann brauchen wir ein Modell, das Energien übersichtlich wiedergibt? Ein Beipiel ist die Besprechung von Halbleitern, die auf VB-Basis kaum sinnvoll beschreibbar sind. Ein anderes Beispiel ist die Beziehung zwischen der Farbe eines Stoffes und seiner Elektronenstruktur (warum ist Hämoglobin rot? Was lässt sich daraus schließen, wenn es bei der Wechselwirkung mit CO oder NO seine Farbe ändert?).
Für die meisten Elemente ist in den Lehrbüchern ein „Atomradius“ angegeben. Bei den nichtmetallischen Elementen handelt es sich dabei in der Regel um den kovalenten Einfachbindungsradius. Dieser ist definiert als die Hälfte des Atomabstandes in einer homonuklearen Einfachbindung. Beispiele: die Atomabstände in H2 und Cl2 betragen 74 und 198 pm. Die kovalenten Radien sind dann 37 und 99 pm. Mit Hilfe dieser Radien können Erwartungswerte für heteronukleare Bindungen errechnet werden. Für HCl ergibt sich so 136 pm (exp. 127 pm).
Auch eine freiwillig ablaufende Reaktion bedarf oft der Zündung – es muss Aktivierungsenergie zugeführt werden. Bei einer Radikalkettenreaktion besteht die Aktivierung in der Bildung erster Radikale. Bei der Chlorknallgasreaktion gelingt dies mit Licht, das die Bindungsdissoziationsenergie des Cl2-Moleküls zur Verfügung stellt. Ein Katalysator senkt die Aktivierungsbarriere.
Versuch 15-1: H2 brennt in Cl2 • Wasserstoff und Chlor reagieren exotherm zu HCl miteinander. Die Reaktion bedarf der Zündung durch eine Flamme.
Versuch 15-3: Chlor-Knallgas • eine Mischung von Wasserstoff und Chlor reagiert nicht spontan, sondern sie ist „metastabil“. Die Aktivierung kann auch durch Lichtenergie erfolgen, allerdings muss die Wellenlänge des verwendeten Lichts einer hinreichend hohen Energie entsprechen.
Die Reaktion von Chlor-Knallgas nach der Zündung kann in einem Energiediagramm dargestellt werden. Die Reaktion läuft erst nach der Zufuhr der Aktivierungsenergie ab. Ea erscheint nicht in der Energiebilanz der Reaktion, die Zustandsgröße ΔH ist von Ea unabhängig.
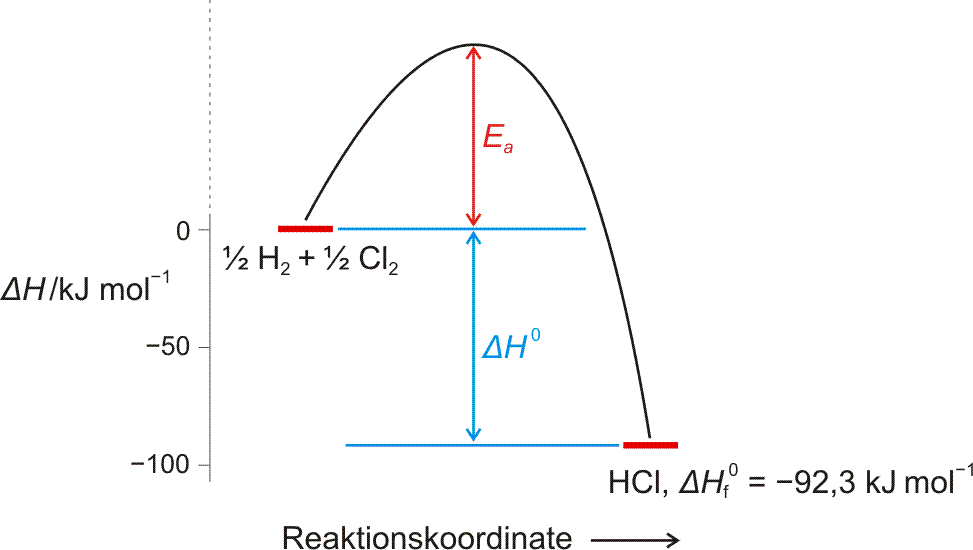
Die Bildung von HCl ist eine besonders übersichtlich verlaufende Reaktion, die es erlaubt, die Notwendigkeit der Aktivierung auf molekularer Ebene zu verstehen. Der erste Schritt der Reaktion besteht im Bruch von Cl-Cl-Bindungen, die schwächer als H-H-Bindungen sind (siehe unten). Die gelbe Farbe von Chlorgas ist bereits ein Hinweis, dass Chlormoleküle auch durch Licht angeregt werden können. Der Zerfall eines Chlormoleküls in zwei Chloratome startet eine Radikalkettenreaktion. Im folgenden Schema ist Gleichung (1) der Kettenstart, der allgemein dadurch charakterisiert ist, dass auf der rechten Seite der Gleichung mehr Radikale, das sind Atome oder Moleküle mit einem ungepaarten Elektron, als auf der linken Seite vorliegen – es entstehen Radikale. Bei den anschließenden Kettenfortpflanzungsreaktionen bleibt die Zahl der Radikale gleich; jeder der beiden Schritte (2a) und (2b) erzeugt Produkt aus einem Eduktmolekül, zugleich bleibt die Zahl der Radikale unverändert. Erst in der Kettenabbruchreaktion (3) verschwinden Radikale und die Kettenreaktion kommt zum Erliegen.
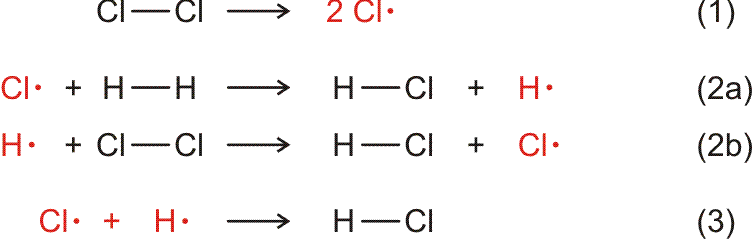
Die Energie, die zum Bruch einer kovalenten Bindung aufgewandt werden muss, ist die Bindungsdissoziationsenergie. Für die bisher behandelten Elemente wird gefunden: D/kJ mol−1: H2 432, N2 942, O2 494, Cl2 240. Man beachte eine gewisse Korrelation mit der Bindungsordnung. Die Werte zeigen, dass ein Chlor-Molekül leichter als ein Wasserstoffmolekül in die Atome gespalten werden kann. Vergleicht man die Energien im Spektrum des sichtbaren Lichts (E/kJ mol−1: 300 bei 400 nm, 240 bei 500 nm, 200 bei 600 nm, 170 bei 700 nm, 150 bei 800 nm), so wird klar, warum blaues Licht den Schritt (1) der Chlorknallgasreaktion einleiten kann, nicht aber rotes; außerdem wird deutlich, warum die Aktivierung nicht in der Spaltung eines H2-Moleküls besteht.
Versuch 5-14: H2 unterhält die Verbrennung einer Kerze nicht, brennt selbst aber
Versuch 5-19: Döbereiner-Feuerzeug
Die Reaktion
2 H2 + O2 → 2 H2O ΔrH° = 2 × ΔfH°(H2O) = −572 kJ mol−!
ist stark exotherm, aber auch sie bedarf der Aktivierung, im Versuch durch eine Kerzenflamme. Die benötigte Aktivierungsenergie wird drastisch herabgesetzt, wenn Wasserstoff mit Platin in Berührung kommt. Platin ist in der Lage, die H-H-Bindung unter Umgebungsbedingungen zu spalten und so die Verbrennung des Wasserstoffs durch die Bildung von H-Atomen einzuleiten.
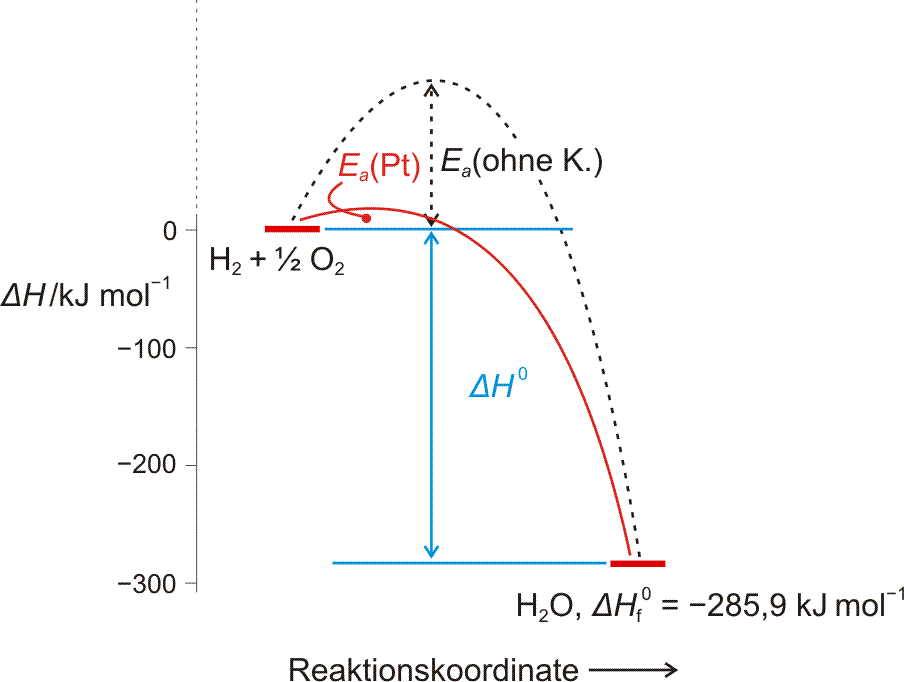
Zur Beschreibung polarer Atombindungen ist das Konzept der Elektronegativität hilfreich. Skalen wie die von Pauling oder Mulliken erlauben es, die Bindungspolarisation quantitativ zu erfassen. Polare kovalente Bindungen sind die Voraussetzung für Wasserstoffbrückenbindungen, die das Zustandsdiagramm des Wassers prägen.
| Mortimer: 8, 9,11 Binnewies: 5.7–5.9 Riedel: 2.2 |
HCl bildet sich bei der Chlorknallgasreaktion in einer deutlich exothermen Reaktion. Bei dieser übersichtlichen Umsetzung, bei der aus 1 Molekül H2 und 1 Molekül Cl2 2 Moleküle HCl entstehen, ist die freiwerdende Energie gleich der Differenz der Bindungsdissoziationsenergie von HCl und dem Mittelwert der Dissoziationsenergien von Wasserstoff- und Chlormolekülen (siehe weiter unten). Warum ist die H–Cl-Bindung unerwartet stark? Dasselbe ist offensichtlich auch bei Wasser zu beobachten.
Auch die Eigenschaften von H2O und HCl, sowie auch die von dessen leichtem Analog HF, lassen sich durch eine einfache Atombindung allein nicht deuten. So neigt HCl außerordentlich stark dazu, auf andere Moleküle ein H+-Ion zu übertragen und dabei ein Cl−-Ion zu bilden (siehe unten im Kapitel „Säure-Base-Reaktionen“). Fluorwasserstoff, HF, ist darüber hinaus nicht, wie aufgrund der kleinen Molekülmasse zu erwarten wäre, bis hin zu tiefen Temperaturen ein Gas, sondern sein Siedepunkt liegt bei 19,5 °C. Dieses Verhalten wird plausibler, wenn für die HF- und HCl-Moleküle eine Polarisierung der Atombindung angenommen wird, wenn der Schwerpunkt des bindenden Elektronenpaars also eher zur Fluor- oder Chlor-Seite hin verschoben wäre. Eine übliche Formulierung für diesen Sachverhalt benutzt die Symbole δ+ und δ− über dem Atomsymbol, um eine Ladung andeuten, die ein Bruchteil der Elementarladung ist. Wie groß aber sind δ+ und δ−? Pauling stellte die erste Skala mit Werten für die Elektronegativität der Atome zusammen, bei der die Fähigkeit eines Atoms, das bindende Elektronenpaar einer kovalenten Bindung zu sich herüberzuziehen, in Zahlen gefasst war. Pauling war aufgefallen, dass eine hierdurch polare Bindung zwischen zwei Atomen eine höhere Bindungsdissoziationsenergie aufweist als erwartet (DHCl = 428 kJ mol−1, ½ DH2 + ½ DCl2 = 336 kJ mol−1). In der Formulierung
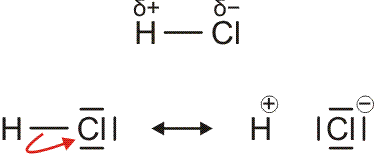
sollte sich eine Stabilisierung der Bindung durch die elektrostatische Wechselwirkung zwischen den Partialladungen ergeben – umso mehr, je größer der Betrag von δ ist. Die untere Formulierung beschreibt denselben Sachverhalt durch zwei mesomere Grenzformeln, die – mit unterschiedlichem Gewicht – zur beobachteten Elektronenverteilung beitragen.
Aus der Stabilisierung kovalenter Bindungen durch Dipolbildung leitete Pauling eine Elektronegativitätsskala ab, die ursprünglich auf den Wert 4.0 für das elektronegativste Element Fluor normiert war. Neben der Pauling-Skala sind weitere Skalen in Gebrauch, zum Beispiel die auf der Ladungsdichte des Atomrumpfes beruhende Allred-Rochow-Skala. Beachten Sie die Trends im Periodensystem.
Elektronegativität (χ) einiger Elemente nach Pauling (P) und Allred-Rochow (AR):
| χ(P) | χ(AR) | |
|---|---|---|
| H | 2,20 | 2,20 |
| Li | 0,98 | 0,97 |
| Be | 1,57 | 1,47 |
| B | 2,04 | 2,01 |
| C | 2,55 | 2,50 |
| N | 3,04 | 3,07 |
| O | 3,44 | 3,50 |
| F | 3,98 | 4,10 |
| Ne | 4,84 | |
| Na | 0,93 | 1,01 |
| Mg | 1,31 | 1,23 |
| Al | 1,61 | 1,47 |
| Si | 1,90 | 1,74 |
| P | 2,19 | 2,06 |
| S | 2,58 | 2,44 |
| Cl | 3,16 | 2,83 |
| Ar | 3,20 | |
| Br | 2,96 | 2,74 |
| I | 2,66 | 2,21 |
Um die Polarität einer Bindung abzuschätzen, wird die Differenz von χ der beiden verbundenen Atome betrachtet. Der Ladungsschwerpunkt ist in einer polaren Bindung zum elektronegativeren Element hin verschoben. Auf der Pauling-Skala entspricht eine Elektronegativitätsdifferenz von 2 einer etwa 50-%-igen Ladungstrennung, das heißt, in der oberen Formel
ist δ ca. 0.5; mit mesomeren Grenzformeln ausgedrückt würde ein Δχ von 2 einer Gleichgewichtung der beiden unteren Formeln, der kovalenten und der ionischen, entsprechen.
Da die Elektronegativität ein Maß für das Bestreben eines Atoms, einem Bindungspartner Elektronendichte zu entziehen, sein soll, erscheint unter den verbreiteten Elektronegativitätsskalen vor allem diejenige von Mulliken als unmittelbar einleuchtend. Er definiert EN-Werte als Mittelwert von 1. Ionisierungsenergie I1 und Elektronenaffinität EA, also als Mittelwert zweier Energiewerte, die das Zufügen und Wegnehmen eines Elektrons wiedergeben.
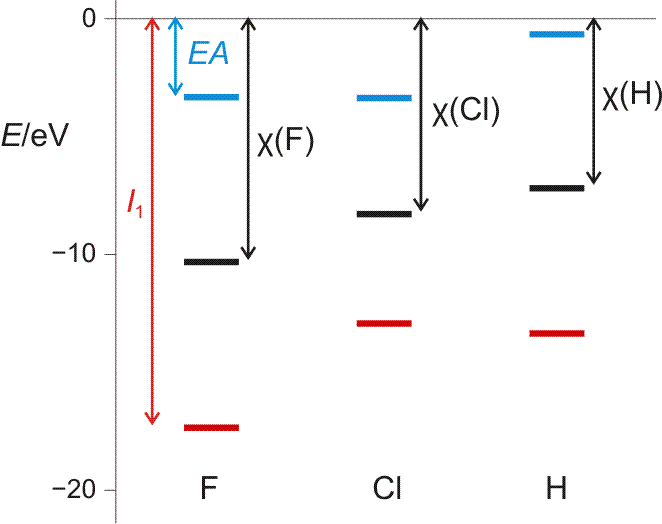
Die Mulliken-Elektronegativität, veranschaulicht mit den I1- und EA-Werten der Atome im Grundzustand (siehe Text).
Die Abbildung stellt den Bezug zu den oben verwendeten Orbitaldiagrammen her, zeigt aber nur das Prinzip des Verfahrens. Soll die Mulliken-Elektronegativität für die Analyse der Bindungssituation eines konkreten Moleküls verwendet werden, so zieht man die χMulliken-Werte für die einzelnen Atome in deren Valenzzustand (s/p-Anteile der Bindungen) heran, wodurch sich oft sehr unterschiedliche Werte für ein und dasselbe Element ergeben (das heißt für uns im Augenblick nur: überinterpretieren Sie nicht kleine Elektronegativitätsunterschiede!).
Das Konzept der polaren Atombindung beschreibt HF als Dipolmolekül (Dipolmoment = Ladung × Abstand der Ladungen). Während die Kräfte zwischen unpolaren Molekülen wie H2, N2 und O2 nur klein sind und diese Stoffe daher Gase mit außerordentlich niedrigem Siedepunkt sind, ziehen sich Dipolmoleküle elektrostatisch an, nachdem sie die günstigste Orientierung zueinander eingenommen haben. Mehr noch als HF ist Wasser das Dipolmolekül schlechthin. Wie HF entsteht auch Wasser in einer stark exothermen Reaktion aus gasförmigen Elementen. Der für ein so kleines Molekül erstaunlich hohe Siedepunkt von 100 °C zeigt an, dass beachtliche zwischenmolekulare (intermolekulare) Kräfte zwischen den H2O-Molekülen wirken müssen.
Das Wassermolekül zeigt sich bei den Methoden zur Strukturbestimmung als gewinkelt. Mit einer Elektronegativitätsdifferenz von ca. 1,3 ist jede der beiden H-O-Bindungen deutlich polar. Die beiden Bindungsdipolmomente addieren sich vektoriell zum Gesamtmoment.
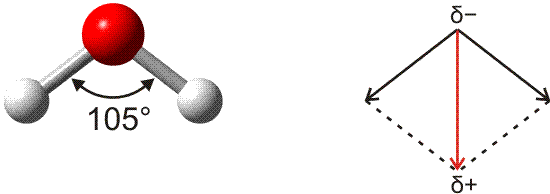
Versuch 13-4: Wasserdipole im elektrischen Feld
Der gewinkelte Aufbau des Wassermoleküls ist aus der Lewisformel unmittelbar ersichtlich, allerdings nicht mit dem Zahlenwert von 105°. Bei einer Bindung von zwei Wasserstoffatomen über die beiden halbbesetzten p-Orbitale eines Sauerstoffatoms sollte vielmehr mit einem 90°-Winkel gerechnet werden. Zu einer korrekteren – halbquantitativen – Aussage gelangt das VSEPR-Modell (valence shell electron pair repulsion, Valenzelektronenpaarabstoßung). In diesem einfachen Modell werden alle Valenzelektronenpaare als gleichwertig behandelt, es wird also nicht zwischen s- und p-Orbitalen unterschieden. Im Sinne des VB-Modells ausgedrückt: es wird von vollständiger Hybridisierung der Valenzorbitale ausgegangen, hier also von sp3-Hybridisierung.
Eine Strukturvorhersage wird nun anhand einiger einfacher Regeln getroffen. Die wichtigste: Die einander abstoßenden Elektronenpaare entfernen sich im Raum möglichst weit voneinander. Die vier Valenzelektronenpaare am Sauerstoffatom sollten damit in die Ecken eines Tetraeders weisen. Mit dieser Struktur ergibt sich für den H-O-H-Winkel zunächst ein Wert von 109,5°. Das Strukturmodell wird durch eine zweite Regel verfeinert: Einsame Elektronenpaare benötigen mehr Raum als bindende Elektronenpaare. Damit wird als Voraussage für den H-O-H-Winkel 109,5° − x erhalten, wodurch der gefundene 105°-Winkel nicht schlecht angenähert wird.
Die Eis-Ih-Struktur zeigt tetraedrische Umgebung um jedes Sauerstoffatom. Die Strukturanalyse zeigt fehlgeordnete Wasserstoffatome entlang der O-O-Verbindungslinien, die eine räumlich und/oder zeitlich gemittelte Situation widerspiegeln:
Die Struktur von Eis1h (gezeichnet in der Raumgruppe P 63/mmc). Die Wasserstoffatome sind „fehlgeordnet“, das heißt, dass die Wahrscheinlichkeit, sie auf der Bindung zum einem der vier nächsten O-Atome anzutreffen, jeweils ½ ist; man beachte 4 × ½ = 2 entsprechend der 2 H-Atome pro O-Atom im Wasser.
Eine reale Wasserstoffatomverteilung könnte so aussehen wie im folgenden Bild (hier dasselbe als bewegtes Bild):
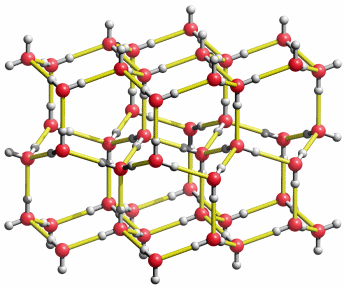
Die Struktur von Eis1h. So könnte eine „Momentaufnahme“ der wirklichen H-Atomverteilung in der mittleren Ebene der vorigen Abbildung aussehen. Durch Platzwechsel entlang der gelb eingezeichneten „Wasserstoffbrückenbindungen“ sind die H-Atome beweglich.
Die in die Eis-Ih-Struktur gelb eingezeichneten Wechselwirkungen sind Wasserstoffbrückenbindungen. Die tetraedrische Umgebung der Sauerstoffatome ist eine Folge dieser Bindungen:
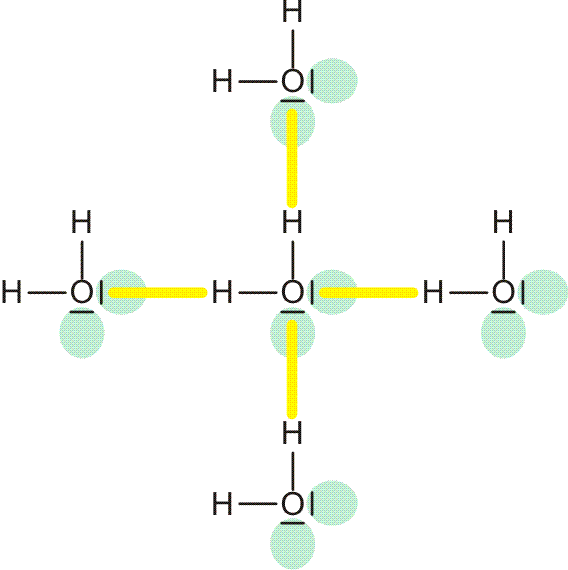
Wasserstoffbrückenbindungen sind durch folgende Merkmale charakterisiert: Das positiv polarisierte H-Atom eines Wasserstoffbrückenbindungsdonors (im folgenden Bild: „hb-Donor“) zeigt eine Wechselwirkung mit einem einsamen Elektronenpaar eines Wasserstoffbrückenbindungsacceptors („hb-Acceptor“). Die entstehende Wasserstoffbrückenbindung ist wie eine kovalente Bindung räumlich gerichtet, hat aber eine viel kleinere Bindungsdissoziationsenergie als diese. Vor allem hängt der Energiebetrag von der Umgebung ab. Ist die Bindung isoliert, ist die Dissoziationsenergie kleiner, ist die Bindung Teil eines Systems hintereinander oder auch cyclisch geschalteter O-H-Vektoren, ist die Energie größer, die Wasserstoffbrückenbindung ist dann Teil eines kooperativen Wasserstoffbrückenbindungssystems.
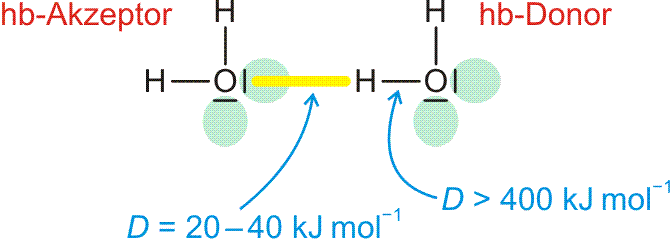
Durch die räumliche Ausrichtung der Bindungen ist in der Eisstruktur eine eher kleine Koordinationszahl realisiert, die Struktur ist im Vergleich zu dicht gepackten Salz- und Metallstrukturen „luftig“. Dies hat wichtige Konsequenzen für die Eigenschaften von Eis und flüssigem Wasser, die auch das Aussehen des Phasendiagramms bestimmen.
Versuch 13-1: Dichteanomalie
Versuch 13-3: Druckabhängigkeit des Schmelzpunktes von Eis
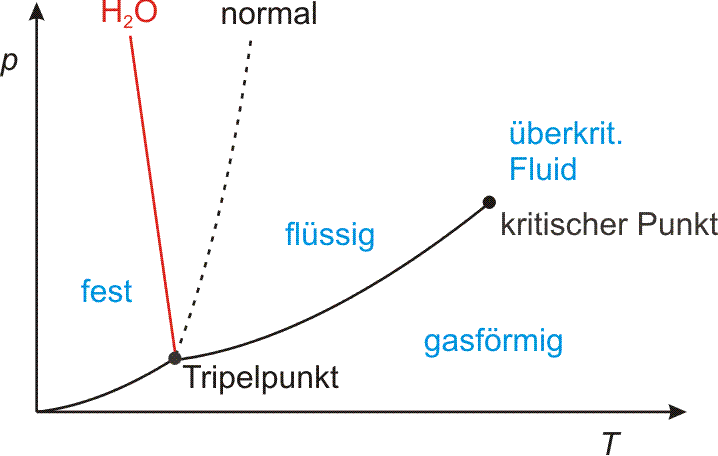
Die Dichteanomalie des Wassers im Druck-Temperatur-Diagramm. Der Tripelpunkt liegt bei 0,01 °C und 0,006 × 105 Pa (Normaldruck: 1,013 × 105 Pa), der kritische Punkt bei 374 °C und 220 × 105 Pa.
Beim Schmelzen von Eis entsteht eine Flüssigkeit, in der die Lücken der Kristallstruktur im Mittel verkleinert sind. Trotz dieser Kontraktion bleibt die Struktur des Eises jedoch ein gutes Modell für die Struktur des flüssigen Wassers. Vor allem das Prinzip, ein Wasserstoffbrückenbindungssystem aufzubauen, führt zur Formulierung eines Clustermodells des Wassers, in dem größere Molekülverbände („Cluster“) nach denselben Regeln organisiert sind wie Ausschnitte aus der Eisstruktur. Ein Beispiel für ein Bauelement, das kooperativ organisiert ist, ist der gezeigte viergliedrige „homodrome“ (gleichsinnig ausgerichtete ) Cyclus:
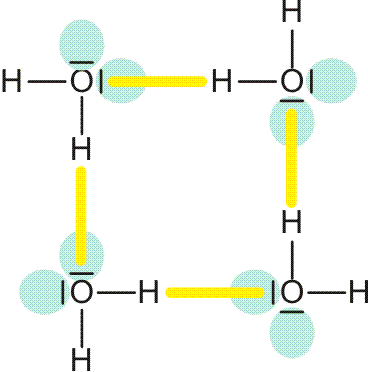
Vor allem, wenn der hb-Acceptor und der hb-Donor ohne H-Atom (demnächst: die konjugierte Base des hb-Donors) gleich sind, werden oft starke Wasserstoffbrückenbindungen gefunden, bei denen der Abstand zwischen Donor- und Acceptoratom kleiner als üblich ist. Bei besonders starken Bindungen ist das H-Atom symmetrisch angeordnet, so dass keine Unterscheidung zwischen Donor und Acceptor möglich ist. In diesem Fall lässt sich die Bindung durch mesomere Grenzstrukturen beschreiben; ein Beispiel ist das HF2−-Ion:
Man vergleiche eine normale und eine symmetrische, starke Wasserstoffbrückenbindung: Statt eines Donor-Acceptor-Abstands von 283 pm in Eis-Ih ist der F···F-Abstand im HF2− kleiner als 230 pm.
Eine Grenzsituation entsteht, wenn der hb-Acceptor eine höheres Bindungsvermögen für das Wasserstoffatom hat als der hb-Donor. Es kommt dann zum Übergang eines Protons auf den Acceptor, wobei das Reaktionsprodukt als wasserstoffbrücken-verbundenes Paar stabil sein kann:
Dieser Situation ist das Kapitel „Säure-Base-Reaktionen“ gewidmet.
Beim Wassermolekül ist die einfache Beschreibung mit Hilfe des VSEPR-Modells erfolgreich: wir nehmen vier gleichwertige Elektronenpaare an, die in der Sprache der VB-Theorie am Sauerstoffatom vier sp3-Hybridorbitale bedeuten. Die tetraedrische Umgebung des Wassermoleküls im Eis ergibt sich so recht zwanglos.
Wie sehen weitergehende Bindungstheorien das Wassermolekül? Wegen der Bedeutung des H2O-Moleküls benutzen wir dieses als Übungsobjekt, um noch einmal die wesentlichen Bindungsmodelle anzuwenden. Zuerst die MO-Theorie, bei der sich für das dreiatomige Wassermolekül neben einer nichtbindenden Wechselwirkung 2e-3z-Bindungen ergeben:
MO-Schema für H2O; Ladung auf O nach Mulliken-Populationsanalyse: −0.60 e (technische Anmerkung: rmp2/aug-cc-pvqz, bei O über α- und β-Spins gemittelt)
Zur Bindung zwischen O- und H-Atomen tragen alle besetzten MOs außer dem energiereichsten bei, das in nichtbindender Anordnung zu den H(1s)-Orbitalen steht. Die Wechselwirkung zwischen den beiden übrigen Sauerstoff-p- mit den H(1s)-Orbitalen führt zur Bindung, zu der auch das O(2s)-Orbital offensichtlich beiträgt (beachten Sie dessen Stabilisierung und die Abweichung von der Kugelgestalt, die es ohne H-Beteiligung hätte).
Etwas irritierend sind die MO-Energien. Wieso ist das nichtbindende Orbital unstabiler als im Atom? Kombinieren Sie zur Erklärung zwei bisher erarbeitete Ergebnisse: (1) Fluor-Valenzorbitale werden deutlich instabiler, wenn das Atom ein Elektron aufnimmt und zum Fluorid-Ion, dem F−-Ion, wird; (2) im polaren H2O-Molekül trägt das O-Atom ungefähr die Ladung −½e.
Wird die s-p-Mischung im Sinne des VB-Modells als Hybridisierung ausgedrückt, so ergeben sich bei der Rechnung O-H-Bindungen, zu denen das Sauerstoffatom zwei Orbitale mit ¼ s-Anteil beiträgt. Dieser Zahlenwert entspricht einer sp3-Hybridisierung, wenn mit gleichwertigen freien Elektronenpaaren formuliert wird (2 × ¼ s-Anteil in den Bindungen lassen ½ s-Anteil für beide freien e-Paare übrig, also ¼ für jedes e-Paar bei Gleichverteilung; eine alternative Sicht auf die freien e-Paare wird bei H2S in Teil B diskutiert). Es wird klar, dass immer dann, wenn die quantitative Behandlung zu Bindungen mit ca. 25% s-Charakter führt, das VSEPR-Modell gut dasteht, bei dem vollständige s-p-Mischung vorausgesetzt wird (Weiters hierzu in Teil B).
Auch die Eigenschaften als Lösungsmittel werden bei Wasser von dessen Dipoleigenschaft bestimmt sowie dem Vermögen, sowohl als Wasserstoffbrückenbindungsdonor wie auch als -acceptor zu fungieren. Molekulardispers gelöste Stoffe lassen sich durch die kolligativen Eigenschaften ihrer wässrigen Lösung charakterisieren. Hierzu zählen die Dampfdruckerniedrigung, Siedepunktserhöhung, Gefrierpunkterniedrigung und der osmotische Druck. Alle diese Größen hängen nur von der Teilchenzahl gelöster Substanz ab, nicht aber von der Art der Teilchen. Die vier Phänomene eignen sich daher zur Molekülmassebestimmung gelöster Stoffe. In der Praxis am weitesten verbreitet sind die Bestimmung der Gefrierpunkterniedrigung (als Standardmethode) und des osmotischen Drucks (vor allem für hochmolekulare Stoffe).
Versuch neu: Gefrierpunktserniedrigung 1-molaler Lösungen von Traubenzucker (Glucose-Monohydrat, C6H12O6·H2O, Mr = 198,17) und Natriumchlorid (Mr = 58,44).
Die Gefrierpunktserniedrigung, die ein gelöster Stoff bewirkt, ist der Molzahl kleinster Teilchen proportional, die in einer bestimmten Masse des Lösungsmittels gelöst ist:
ΔT = Kc · b
b ist die Molalität, die Molzahl an gelöstem Stoff pro kg Lösungsmittel, die Proportionalitätskonstante Kc ist die kryoskopische Konstante, die für Wasser den Wert 1,86 K mol−1 kg hat (eine 1-molale Lösung führt zu 1,86 K Gefrierpunktserniedrigung).
Der Grenzfall der polaren Atombindung bei hoher Elektronegativitätsdifferenz ist die Ionenbindung. Mit der Übertragung des bindenden Elektronenpaars auf das elektronegativere Element geht die räumliche Ausrichtung der Bindung verloren. Salzstrukturen zeigen daher andere Bauprinzipien als Strukturen, die durch kovalente Verknüpfung oder Wasserstoffverbrückung entstehen.
| Mortimer: 7, 20 Binnewies: 4, 7 Riedel: 2.1 |
Die NaCl-Struktur ist eine prototypische Ionenstruktur. Als Bauprinzip fällt die dichte Packung der Kristallbausteine auf, bei der jede Ionensorte oktaederisch von Gegenionen umgeben ist:
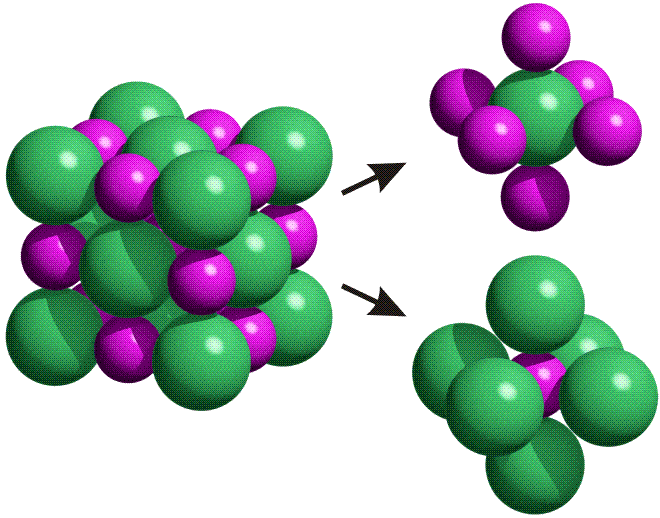
Die Bindung zwischen den Ionen ist weitgehend elektrostatisch. Die energetische Situation erschließt sich durch ein besonders aufschlussreiches Werkzeug, einem „Kreisprozess“. Vor allem die Beobachtung, dass bei der Bildung eines Salzes Ionen mit Edelgaskonfiguration entstehen, wird hierdurch in das richtige Licht gerückt.
Versuch 12-2: Synthese von Natriumchlorid aus den Elementen
Die Bildung eines Ionenkristalls aus den Elementen, die für das Beispiel NaCl mit der Energiebilanz
Na(s) + ½ Cl2(g) → NaCl(s) ΔfH° = −411 kJ mol−1
abläuft, wird für die Formulierung eines Kreisprozesses in Teilschritte zerlegt. So ergeben sich für NaCl die folgenden Schritte mit den entsprechenden Energien:
(a) Die Bildung von einzelnen Na-Atomen aus festem Natrium, die Sublimationsenergie ΔsubH°,
(b) die Bildung von einzelnen Cl-Atomen aus Cl2, die Bindungsdissoziationsenergie D,
(c) die Bildung eines Na+-Ions durch Ionisierung eines Na-Atoms, die 1. Ionisierungsenergie, I1,
(d) die Bildung eines Cl−-Ions durch Zufügen eines Elektrons zu einem Cl-Atom, die Elektronenaffinität EA,
(e) im letzten Schritt dann die Bildung des Ionenkristalls aus Na+- und Cl−-Ionen, die aus unendlicher Entfernung im Gasraum zum Kristall zusammentreten. Dieser Vorgang setzt die Gitterenergie Ug frei.
Die oben angegebene Standardbildungsenthalpie ΔfH° ist die Summe aus diesen Einzelschritten:
ΔfH° = ΔsubH°(Na) + ½ D(Cl2) + I1(Na) + EA(Cl) + Ug
Für NaCl ergeben sich als Zahlenwerte (in kJ mol−1):
−411 = + 108 + 121 + 496 − 356 − 780
Die Energiebilanz zeigt, dass die Bildung der Edelgaskonfigurationen allein Energie benötigt: Der Vorgang
Na(g) + Cl(g) → Na+(g) + Cl−(g)
ist nämlich die Summe der Teilschritte:
Na(g) → Na+(g) + e−
und
Cl(g) + e− → Cl−(g)
Für den gesamten Vorgang ergibt sich:
ΔrH° = I1 + EA = +496 + (−356) = +140 kJ mol−1
Der Bildung von Na+- und Cl−-Ionen aus Na- und Cl-Atomen ist also endotherm. Eine wichtige Schlussfolgerung ist, dass beim Zuführen der Bindungsdissoziationsenergie zu NaCl die Ionenbindung genauso wie die kovalente Bindung unter Bildung von Atomen gespalten wird – die beiden wichtigen Grundtypen der chemischen Bindung verhalten sich also bei Zufuhr der Bindungsdissoziationsenergie gleich, beide werden homolytisch gespalten.
Der größte Einzelbetrag im Kreisprozess ist die Abgabe der Gitterenergie, also der Energie, die frei wird, wenn Ionen sich aus unendlicher Entfernung zu einem Ionengitter zusammenfinden. Zum stark exothermen Verlauf vieler Bildungsreaktionen von ionisch aufgebauten Stoffen aus den Elementen leistet die Gitterenergie meist den größten Beitrag.
Der erste Schritt beim Aufbau eines Ionenkristalls ist die Bildung eines Ionenpaares Na+Cl−. Der energetische Beitrag dieses ersten Schrittes zur Gitterenergie lässt sich berechnen. Bei der Annäherung von Na+- und Cl−-Ionen
• sinkt das zu r−1 proportionale Coulomb-Potential
| UC, Ionenpaar = | −z+ z− e2 |
| 4 π ε0 r |
allmählich (im Bild blau gestrichelt),
• steigt ein abstoßendes Potential schnell an (im Bild grün dargestellt; bei NaCl proportional zu ca. r−8), wenn der Abstand der Ionen einen Grenzwert unterschreitet, unterhalb dessen die Elektronenhüllen der Ionen in größerem Umfang einander durchdringen würden:
Der Zahlenwert der potentiellen Energie im Gleichgewichtsabstand (Ug im Bild) wird näherungsweise durch den Coulomb-Term allein bestimmt (UC im Bild). Für Na+Cl− (Kernabstand im Ionenpaar = 238 pm) ergibt eine solche Betrachtung −584 kJ mol−1. Verglichen mit der Gitterenergie von −780 kJ mol−1 zeigt sich, dass die Bildung des Ionenpaars Na+Cl− diesen Betrag zu einem großen Teil bewirkt. Die Berechnung erklärt das Verhalten von Salzen beim Verdampfen oder beim Auflösen in schlecht solvatisierenden Lösungsmitteln – hier liegen keine einzelnen Ionen vor, sondern Ionenpaare oder höhere Assoziate.
Um in ähnlicher Weise den Coulomb-Anteil UC an der Gitterenergie berechnen zu können, ist die Umgebung der Ionen zu betrachten. Für ein Na+-Ion im NaCl besteht diese Umgebung aus:
6 Cl− im Abstand d,
12 Na+ im Abstand √2 d,
8 Cl− im Abstand √3 d,
6 Na+ im Abstand √4 d = 2 d,
24 Cl− im Abstand √5 d, usw.
Die Coulomb-Energie der NaCl-Struktur ergibt sich als Summe der Einzelbeiträge, wobei die Häufigkeit der Abstände zu beachten ist; für r werden die entsprechenden Abstände als Vielfache von d eingesetzt, dem gefundenen Abstand zwischen den Ionen (im NaCl-Kristall 283 pm); z+ und z−, die Beträge der Ionenladungen (= 1 für NaCl) ist angegeben, da die Beziehung auch für andere Verbindungen mit diesem Strukturtyp gilt; anziehende Energiebeiträge werden mit negativem Vorzeichen eingesetzt, abstoßende positiv. Für 1 mol Salz gilt:
| UC = − | NA z+ z− e2 | (6 − 12/√2 + 8/√3 − 6/√4 + 24/√5 …) |
| 4 π ε0 r |
Der Klammerausdruck, der Grenzwert für unendlich viele Summanden, ist die Madelung-Konstante A, die nur vom Aufbau der Struktur abhängt. Für den NaCl-Strukturtyp ist der Zahlenwert 1,7476. Er gibt an, um welchen Faktor der Ionenkristall stabiler ist als ein gedachtes Ionenpaar, bei dem der Ionenabstand derselbe ist wie im Kristall. Die Gleichung für UC vereinfacht sich damit zu:
| UC = − | NA A z+ z− e2 |
| 4 π ε0 r |
Der vernachlässigte Abstoßungsterm trägt zu ca. 10 % zur Gitterenergie bei, um den die nur als UC abgeschätzten Werte vom Betrag her zu hoch ausfallen. Im Laufe der Zeit wurden verschiedene Näherungsverfahren beschrieben, um Ug präziser berechnen zu können. Verbreitet ist die Born-Landé-Formel:
| UC = − | NA A z+ z− e2 | (1 − 1/n) |
| 4 π ε0 r |
Hier wird ein Korrekturterm 1/n benutzt, der von der Elektronenkonfiguration der beteiligten Ionen abhängt. Er kann die folgenden Zahlenwerte annehmen:
| Konfiguration | Beispiel | n |
|---|---|---|
| [He] | H−, Li+ | 5 |
| [Ne] | O2−, F−, Na+, Mg2+ | 7 |
| [Ar], 3d10 | Cl−, K+, Cu+ | 9 |
| [Kr], 4d10 | Br−, Sr2+, Cd2+ | 10 |
| [Xe], 5d10 | I−, Cs+, Ba2+ | 12 |
Für NaCl ergibt sich im Rahmen der Born-Landé-Näherung mit NA = 6,022 ×1023 mol−1, A = 1,7476 für NaCl, e = 1,602 × 10−19 C, n = 8 als Mittelwert von 7 und 9, ε0 = 8,854 ×10−12 F m−1 = 8,854 ×10−12 C2 J−1 m−1, 2,83 × 10−10 m Ionenabstand in NaCl:
| UBorn = − | 6,022 ×1023 mol−1 × 1,7476 × (1,602 × 10−19 C)2 × (1 − 1/8) | = 751 kJ mol−1 |
| 4 π 8,854 ×10−12 C2 J−1 m−1 × 2,83 × 10−10 m |
Gitterenergien lassen sich nicht direkt messen, können als Zustandsgrößen aber indirekt bestimmt werden. Die im Kreisprozess verwendete Gleichung lässt sich zum Beispiel nach Ug auflösen; alle anderen Summanden können experimentell bestimmt werden. Man vergleiche den so erhaltenen Wert von −780 kJ mol−1 mit dem Wert von −751 kJ mol−1 nach der Born-Landé-Formel.
Werden verschiedene Verbindungen gleicher Struktur betrachtet, so ergibt sich aus der Betrachtung des Coulomb-Anteils eine Vergrößerung der Gitterenergie bei
• einer Erhöhung der Ladung der Ionen; man vergleiche Na+Cl− und Ba2+O2−:
NaCl (d = 283 pm), z+ = z− = 1: Ug = −780 kJ mol−1,
BaO (d = 276 pm), z+ = z− = 2: Ug = −3128 kJ mol−1,
Es folgt: Ug(BaO) ≈ 4 Ug(NaCl).
• einer Verkleinerung des Abstandes der Ionen; man vergleiche die Gitterenergie von BaO (d = 276 pm) mit dem Wert für MgO (d = 212 pm) von −3936 kJ mol−1.
Gitterenergien sind ein Maß für die Stärke der Bindung in einem Ionenkristall, sie sind daher zum Beispiel in der Reihe NaCl, BaO, MgO mit anderen physikalischen Eigenschaften wie dem Schmelzpunkt (800, 1925, 2642 °C) und der Härte (Ritzhärte nach Mohs: 2,5; 3,3; 6) verbunden.
Versuch 12-3: NaCl-Lösung leitet den Strom
Der kryoskopische Versuch hatte die Dissoziation von NaCl in Wasser gezeigt. Die Umgebung der Ionen im Kristall ist dabei Vorbild für die sehr ähnliche Umgebung in Lösung. Beim Auflösen des Salzes werden die Ionen von Wassermolekülen in der gleichen Weise umlagert wie sie im Kristall Gegenionen als Nachbarn hatten. So geht das NaCl6-Fragment des Kristalls in ein [Na(H2O)6]+-Ion in Lösung über. Das Natrium-Ion wird hydratisiert (allgemein: solvatisiert), es bildet sich ein Aquakomplex, hier das Hexaaquanatrium-Ion. An die Stelle der Chlorid-Gegenionen sind Wasserdipole getreten, die sich mit ihrem negativen Ende an die Kationen und mit der Gegenseite an die Anionen anlagern.
Die starke Analogie zwischen Ionenkristall und Lösung macht sich vor allem in der Energiebilanz bemerkbar. Die analog zur Gitterenergie definierte Hydratationsenthalpie (die bei der Hydratation von 1 mol Ionen freiwerdende Energie) hat annähernd denselben Betrag wie die Gitterenergie. Für NaCl (Gitterenergie −780 kJ mol−1) beträgt die Differenz zwischen Gitterenergie und Hydratationsenthalpie, die Lösungsenthalpie, nur 3,6 kJ mol−1.
Versuch 3-11: NH4NO3 löst sich endotherm
Versuch 12-8: CaCl2 löst sich exotherm, das Hexahydrat endotherm
Bei einem endotherm in Lösung gehenden Salz wie Natriumchlorid ist die Gitterenergie größer als die Solvatationsenthalpie. Hier stellt sich natürlich die Frage, warum sich NaCl überhaupt in Wasser auflöst?
Die bisher benutzte Näherung: „Exotherme Reaktionen laufen (spätestens nach Aktivierung) freiwillig ab, endotherme Reaktionen müssen durch laufende Energiezufuhr erzwungen werden“, gilt nur für Reaktionen mit starker Wärmetönung (vergleiche die Standardbildungsenthalpie von NaCl von −411 kJ mol−1 mit der Lösungsenthalpie von +3,6 kJ mol−1!). Maßgeblich für den freiwilligen Ablauf einer chemischen Reaktion, hier des Lösevorgangs, ist jedoch nicht die Reaktionsenthalpie. Vielmehr läuft eine chemische Reaktion (spätestens nach Aktivierung) freiwillig ab, wenn die Unordnung des Weltalls (als Summe der Unordnung des betrachteten Systems und der Unordnung der Umgebung) größer wird.
Zur quantitativen Beschreibung dient die Gibbs-Helmholtz-Beziehung:
ΔG = ΔH − T ΔS
ΔG ist die Änderung einer weiteren Zustandsgröße des betrachteten Systems, der freien Enthalpie G. ΔG ist mit der Änderung der Unordnung im Weltall verknüpft. Eine Vergrößerung der Gesamtentropie, der Bedingung für eine freiwillig ablaufende Reaktion, geht einher mit:
ΔG < 0
Die Gibbs-Helmholtz-Beziehung zerlegt die gesamte Entropieänderung in zwei Teile: der Entropieänderung in der Umgebung, die mit ΔH verknüpft ist (siehe oben) und der Entropieänderung im betrachteten System, ΔS.
NaCl löst sich in Wasser trotz des endothermen Reaktionsverlaufs auf, da aufgrund eines positiven T ΔS-Terms ΔG negativ wird. T ΔS ist positiv, wenn ΔS positiv ist, wenn also die Entropie des Systems größer wird. Für die Auflösung von NaCl ist dies einsichtig, da das geordnete Kristallgitter zerstört wird, und frei in der Lösung sich bewegende Ionen erzeugt werden.
ΔG, ΔH und T ΔS haben für einige Salze die folgenden Werte (in kJ mol−1):
| ΔH | T ΔS | ΔG | |
|---|---|---|---|
| NaCl | +3,6 | +12,8 | −9,2 |
| AgF | −20,3 | −5,8 | −14,5 |
| NH4Cl | +15,1 | +21,8 | −6,7 |
| MgCl2 | −155,0 | −29,0 | −126,0 |
Für Silberfluorid, AgF, und Magnesiumchlorid, MgCl2, ergeben sich negative T ΔS-Terme. Die Unordnung des Salz/Wasser-Systems nimmt beim Auflösen dieser Salze ab. Dies ist möglich, da beim Lösen zwar das geordnete Kristallgitter zerstört wird, auf der anderen Seite aber Wassermoleküle durch das Einfügen in eine Solvathülle geordnet werden. Erwartungsgemäß ist dieser Effekt bei MgCl2 mit seinem kleinen Mg2+-Kation hoher Ladungsdichte groß.
Ebenso wie sich die Reaktionsenthalpie ΔrH° als Summe von Standardbildungsenthalpien der einzelnen Reaktanden berechnen lässt, kann auch die freie Standardenthalpie ΔrG° einer Reaktion als Summe freier Standardbildungsenthalpien ΔfG° berechnet werden. Die erhaltenen ΔG°-Werte gelten für Standardbedingungen bezüglich Druck und Temperatur, aber auch in Bezug auf die Konzentrationen der Reaktanden. Aus der Darstellung von ΔG als maximaler Nutzarbeit einer Reaktion (vergleiche Lehrbücher der Thermodynamik) lässt sich eine Beziehung zwischen ΔG und ΔG° für andere Konzentrationen der Reaktanden herleiten. Für eine allgemeine Reaktion
a A + b B → x X + y Y
gilt im Falle verdünnter Lösungen:
| ΔG = ΔG° + R T ln | [X]x [Y]y |
| [A]a [B]b |
Reaktionssteuerung, Katalyse, vernetzte Gleichgewichte – die Behandlung des chemischen Gleichgewichts ist Voraussetzung für ein tieferes Verständnis chemischer Prozesse sowohl solcher der Technik als auch solche in der belebten Natur. Dreh- und Angelpunkt in diesem und den folgenden Kapiteln: das Massenwirkungsgesetz
| Mortimer: 15.8, 16, 19 Binnewies: 9, 13.4 Riedel: 3.5 |
Bei der Reaktion von Wasserstoff mit Iod entsteht Iodwasserstoff:
H2 + I2 → 2 HI
Die Umsetzung verläuft nicht vollständig, sondern es entsteht ein Gemisch, das neben HI noch H2 und I2 enthält. So reagieren bei einer Temperatur von 490 °C in einem Volumen von 1 L 1 mmol H2 (1 mmol = 10−3 mol) und 1 mmol I2 zu einem Gemisch aus 1,544 mmol HI, 0,228 mmol H2 und 0,228 mmol I2. Werden 2 mmol HI auf die gleiche Temperatur von 490 °C erhitzt, so findet die umgekehrte Reaktion statt:
2 HI → H2 + I2
Aus 2 mmol HI bildet sich nach einiger Zeit ebenfalls ein Gemisch aus 1,544 mmol HI, 0,228 mmol H2 und 0,228 mmol I2. Es kommt also auf beiden Wegen zur Ausbildung eines Gleichgewichtszustands, der durch eine bestimmte Zusammensetzung des Stoffgemisches charakterisiert ist – es hat sich ein chemisches Gleichgewicht eingestellt:
H2 + I2 ⇄ 2 HI
Ein chemisches Gleichgewicht entspricht keinem Ruhezustand, in dem sich die beteiligten Moleküle – im Beispiel HI, H2 und I2 – ohne weitere Veränderung durch den Gasraum bewegen. Es liegt vielmehr ein dynamisches Gleichgewicht vor, in dem die Geschwindigkeiten der Hinreaktion und der Rückreaktion gleich sind.
Zur quantitativen Beschreibung der Gleichgewichtslage wird von einer allgemein formulierten Reaktion ausgegangen:
a A + b B ⇄ x X + y Y
Für die Änderung der freien Enthalpie der Reaktion gilt im Gleichgewichtszustand:
ΔG = 0
Mit den Gleichgewichtskonzentrationen der beteiligten Reaktanden gilt dann:
| 0 = ΔG° + R T ln | [X]x[Y]y |
| [A]a[B]b |
[A], [B], [X] und [Y] sind Vielfache einer Standardkonzentration von 1 mol L−1; ein analoger Ausdruck kann mit Vielfachen eines Standarddruckes von 1 atm = 1,013 bar = 101,3 kPa formuliert werden. Liegt zum Beispiel die Komponente A in der Konzentration c(A) = 0,05 mol L−1 vor, so gilt: [A] = 0,05. (Ein Grund für diese scheinbar umständliche Art, Konzentrationen einzusetzen, liegt in der Verwendung logarithmischer Beziehungen: ln(0,3 mol L−1) ist nicht definiert, wohl aber ln(0,3). Es wäre übrigens demnächst nötig, dass Sie mit Logarithmen klarkommen.)
Nach Umstellen ergibt sich:
ΔG° = −RT lnK
K ist die Gleichgewichtskonstante der Reaktion. Bei 25 °C = 298,15 K gilt:
ΔG° = −(8,314 × 10−3 kJ mol−1 K−1 × 298,15 K) ln K
= −2,479 kJ mol−1 × ln K
K wird mit dem Index „c“ versehen, wenn die Konstante auf Konzentrationen beruht und mit einem Index „p“, wenn der Berechnung Drücke zugrunde liegen. Die Beziehungen
| [X]x[Y]y | = Kc |
| [A]a[B]b |
und
| px(X) py(Y) | = Kp |
| pa(A) pb(B) |
stellen das Massenwirkungsgesetz (MWG) dar. Der Betrag von K gibt die Zusammensetzung des betreffenden Stoffgemisches nach der Einstellung des Gleichgewichtes an. Für das Beispiel der oben beschriebenen Reaktion von Wasserstoff und Iod zu Iodwasserstoff errechnet sich Kc für eine Temperatur von 490 °C aus den oben angegebenen Mengen zu:
| [HI]2 | = Kc = | (1,544)2 | = 45,9 |
| [H2] [I2] | 0,228 · 0,228 |
Das Massenwirkungsgesetz gilt für homogene Gleichgewichte, bei denen alle an der Reaktion beteiligten Stoffe in derselben Phase vorliegen. Bei hinreichend hohen Temperaturen gilt dies zum Beispiel für die technisch wichtigen Gleichgewichte:
3 H2 + N2 ⇄ 2 NH3
und
2 SO2 + O2 ⇄ 2 SO3
Für die entsprechenden Gleichgewichtskonstanten gilt:
| [NH3]2 | = Kc |
| [H2]3 [N2] |
und
| [SO3]2 | = Kc |
| [SO2]2 [O2] |
Bei heterogenen Gleichgewichten, an denen Stoffe in mehreren Phasen beteiligt sind, kann das MWG für homogene Teilgleichgewichte formuliert werden. So kann bei den folgenden Reaktionen für die Gasphase mit Konzentrationen oder mit Partialdrücken formuliert werden:
C(s) + O2(g) ⇄ CO2(g) Kc = [CO2] [O2]−1
C(s) + CO2(g) ⇄ 2 CO(g) Kp = p2(CO) · p−1(CO2)
CaCO3(s) ⇄ CaO(s) + CO2(g) Kp = p(CO2)
Solange bei diesen Reaktionen noch Feststoff vorliegt, kann es im Gasraum zur Gleichgewichtseinstellung kommen, gleichgültig, wieviel Feststoff genau vorhanden ist.
Einige der Grundprinzipien bei der Behandlung chemischer Gleichgewichte werden am Beispiel der beiden Stickstoffoxide NO und NO2 behandelt. Diese werden daher zuerst kurz vorgestellt.
Radikale sind meist so reaktiv, dass sie nicht als handhabbare Verbindungen vorkommen. Neben dem Diradikal Sauerstoff gilt dies nicht für die beiden Stickstoffoxide NO und NO2, bei denen es sich um 11- bzw. 17-Valenzelektronenteilchen handelt. Beim Aufstellen ihrer Lewis-Formeln wird die Oktettregel beachtet. Wegen der vier Valenzorbitale eines Elementes des p-Blocks werden maximal acht Valenzelektronen formuliert. Bei Stickstoffmonoxid, NO, führt dies in vielen Lehrbüchern zu den beiden mesomeren Grenzformeln rechts, aus denen allerdings die Bindungsordnung von 2,5 nicht hervorgeht, die im MO-Schema erhalten wird. Die linke Grenzformel zeigt daher eine 3-Elektronen-2-Zentren-Formulierung, die vor einem halben Jahrhundert von Pauling vorgeschlagen wurde. Die ausgefüllten Kreise bedeuten hier Elektronen mit Mehrheitsspin (die Pfeile nach oben in den Schemata, alpha-Spin), der offenen Kreis den Minderheitsspin (Pfeile nach unten, beta-Spin; in der Literatur finden Sie anstelle des offenen Kreisen auch schon mal ein Kreuz – allerdings sehr uneinheitlich, es sind weitere Schreibweisen im Umlauf). In dieser Formulierung wird die Bindungsordnung korrekt dargestellt. Wir schauen uns an, wie man die Formel aus dem MO-Schema von NO herleiten kann.
Zuvor eine kurze Anmerkung: Die äußerst rechte Formel weist Formalladungen auf. Diese sind in der Lewis-Formel nicht unbedingt notwendig, helfen aber am Anfang, das Gewicht einer Grenzformel einzuschätzen. Um die Formalladung auf einem Atom zu bestimmen, werden alle Bindungen in Gedanken homolytisch gespalten. Anschließend wird die Elektronenzahl mit der Valenzelektronenzahl des Atoms verglichen. Da im Beispiel für das N-Atom 6 Elektronen gezählt werden, ergibt sich ein Überschuss von 1 Elektron – die Formalladung ist 1−. Sind, wie bei NO, die Elektronegativitätsunterschiede zwischen den beteiligten Atomen gering, gilt die Regel: je mehr Formalladungen, desto geringer das Gewicht der Formel. Das Gewicht der Formel erniedrigt sich weiter, wenn positive und negative Ladungen entgegengesetzt zur Elektronegativität verteilt werden.
Nun zum MO-Schema. Die Schemata von NO und O2 weisen große Ähnlichkeiten auf, bei NO steht lediglich für die antibindenden Orbitale mit π-Symmetrie nur eines statt zwei Elektronen wie bei Sauerstoff zur Verfügung; es zeigt sich, dass das ungepaarte Elektron tatsächlich nennenswert über das Molekül delokalisiert ist, wenn auch seine Aufenthaltswahrscheinlichkeit am Stickstoffatom etwas größer ist:
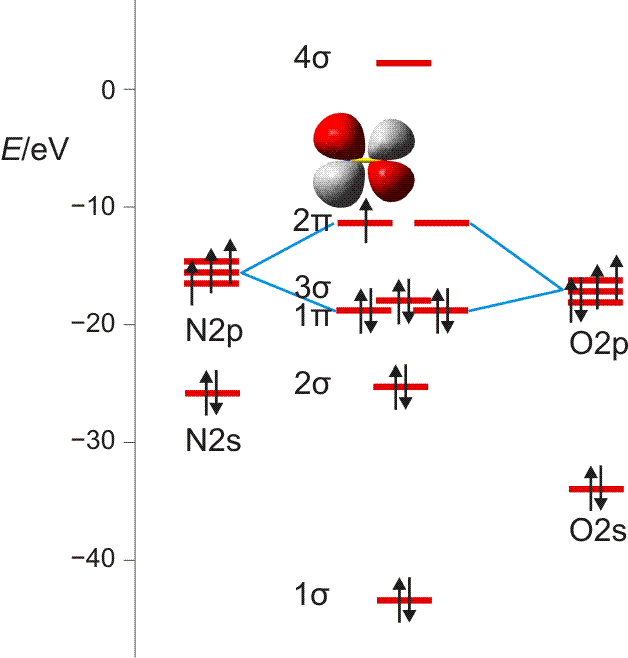
MO-Diagramm von NO im Dublett-Grundzustand; das mit dem ungepaarten Elektron besetzte Orbital ist eingezeichnet, die übrigen Orbitale entsprechen denjenigen im N2-Molekül (techn. Anm.: ub3lyp/cc-pvqz//ump2/aug-cc-pvqz; N-O 114,455 pm; α- und β-Spin-Energien gemittelt, 1π-Energien gemittelt; 2π0β bei der Energie von 2π1α eingezeichnet).
Entsprechend der Stellung des NO zwischen Stickstoff (Bindungsordnung = 3) und Sauerstoff (BO = 2) ergibt sich die Bindungsordnung im NO aus dem MO-Schema zu 2,5.
NO, das sowohl als technisches Zwischenprodukt vor allem bei der Düngemittelherstellung als auch als gefäßerweiterndes Hormon große Bedeutung hat, hat eine positive Standardbildungsenthalpie:
½ N2 + ½ O2 → NO ΔfH° = +90,4 kJ mol−1
Der Zerfall von NO in die Elemente ist also mit −90,4 kJ mol−1 exotherm. Dass NO bei Raumtemperatur gehandhabt werden kann, zeigt, dass es metastabil ist.
Versuch 23-19: NO-Synthese im Lichtbogen • in einen luftgefüllten 2-L-Kolben lässt man über einige Minuten elektrische Entladungen einwirken. Der Gasraum wird schnell braunrot, da sich NO2 bildet. Wird das Gas ausgeleitet, so verlaufen Nachweisreaktionen für NO/NO2 positiv, zum Beispiel entsteht mit „Lunges Reagens“ eine typische Rotfärbung durch die Bildung eines Azofarbstoffs (näheres in den Praktika beim Nitrit/Nitrat-Nachweis).
Sowohl die Bildungweise aus den Elementen als auch die Metastabilität von NO spiegeln eine Reihe von fundamentalen Regeln wider. Die zweiatomigen Moleküle beider Ausgangsstoffe, N2 und O2, sowie NO selbst sind durch Mehrfachbindungen mit hoher Bindungsdissoziationsenergie charakterisiert. Dies zieht eine hohe Aktivierungsenergie nach sich, wenn die beiden Elemente in einer unkatalysierten Gasreaktion miteinander umgesetzt werden sollen. Da die Reaktion endotherm ist, muss ständig weiter Energie zugeführt werden. Da das Produkt metastabil ist, darf nach der Reaktion die Aktivierungsenergie für die Rückreaktion nicht zur Verfügung stehen – eine Bedingung, die einfach zu erfüllen ist, da auch der NO-Zerfall aus dem gleichen Grund wie bei N2 und O2 hohe Aktivierungsschwellen aufweist, solange er unkatalysiert erfolgt.
Stickstoffdioxid, NO2, ist ebenfalls ein Radikal. Um dessen Valenzelektronenanordnung durch Lewis-Formeln zu beschreiben, sind mehrere mesomere Grenzformeln notwendig. Hier die in Lehrbüchern verbreiteten, an deren Stelle auch hier 3-Elektronen-2-Zentren-Formulierungen eine gute Alternative wären, die hier aber nicht gezeichnet sind:
Diesmal gibt es keine Grenzformel, der aufgrund der genannten Regeln ein besonders niedriges Gewicht zuzuschreiben wäre. Die beiden rechten Formeln sollten wegen des Auftretens von Formalladungen ein geringeres Gewicht haben, allerdings sind die Ladungen vom Vorzeichen her im Einklang mit den Elektronegativitätswerten. Insgesamt erscheint die Annahme sinnvoll, das ungepaarte Elektron sei ziemlich gleichmäßig über das dreiatomige Molekül delokalisiert. Im Fall des NO2 wird die Lewis-Formulierung durch eine quantitative MO-Rechnung recht gut bestätigt. Am energiereichsten, einfach besetzten Orbital (dem „SOMO“ von singly occupied molecular orbital) sind auch nach Anwendung der MO-Methode Atomorbitale aller drei Atome ungefähr gleich beteiligt (erkennen Sie welche?):
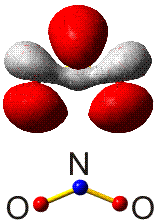
Auch NO2 ist metastabil in Bezug auf den Zerfall in die Elemente, die Standardbildungsenthalpie beträgt nun jedoch nur +33,8 kJ mol−1. Die für ein Radikal in Abwesenheit anderer Reaktionspartner erwartete Stabilisierung durch Dimerisierung ist bei NO nur bei tiefen Temperaturen zu beobachten. Das braune Gas NO2 hingegen liegt schon bei Raumtemperatur zu einem Teil als farbloses Dimer vor, dem Distickstofftetroxid:
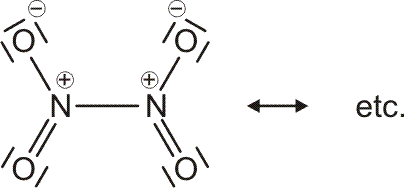
Dass diese diamagnetische Verbindung nicht stabiler ist, hängt mit einer untypisch schwachen N-N-Bindung zusammen. Eine ausführliche Diskussion wird hier nicht gegeben (es hängt damit zusammen, dass die im NO2-MO-Bild gezeigte Delokalisierung im SOMO auch nach der Spinpaarung im Dimer fortbesteht, so dass die Bindungselektronen in geringerem Umfang zwischen den verbundenen Atomrümpfen lokalisiert sind als es bei einer kovalenten Bindung sonst üblich ist). Man beachte, dass die Reaktionsenthalpie für den Zerfall von N2O4 der Bindungsdissoziationsenthalpie der N-N-Bindung entspricht; der Zahlenwert ist ungewöhnlich niedrig:
N2O4 → 2 NO2 ΔrH° = 57,9 kJ mol−1
Die Gleichgewichtslage chemischer Reaktionen kann durch eine Reihe von Parametern beeinflusst werden:
• Durch Änderung der Konzentrationen bzw. der Partialdrücke der Reaktionsteilnehmer;
• durch Temperaturänderung;
• bei Reaktionen mit Änderung der Gesamt-Molzahl durch Änderung der Gesamtkonzentration (bei Gasen: des Gesamtdruckes).
Ein Beispiel ist das oben formulierte NO2/N2O4-Gleichgewicht.
Versuch 16-5: NO2-N2O4-Gleichgewicht
Die Temperatur- und Druckabhängigkeit der Gleichgewichtslage wird qualitativ durch das – für eine naturwissenschaftliche Regel ungewöhnlich anthropomorphe – „Prinzip vom kleinsten Zwang“ (Prinzip von Le Chatelier) beschrieben: „Übt man auf ein System im Gleichgewicht durch Konzentrationsänderung (Druckänderung) oder Temperaturänderung einen Zwang aus, so verschiebt sich das Gleichgewicht so, dass der Zwang vermindert wird“. Für das NO2/N2O4-Gleichgewicht würde folgendermaßen argumentiert: Wird die Temperatur erhöht, so versucht das System, der Temperaturerhöhung durch Wärmeverbrauch auszuweichen. Dies geschieht bei der endothermen Stickstoffdioxid-Bildung durch vermehrte Hinreaktion: Der Zerfall von N2O4 ist endotherm, es wird Wärme verbraucht und damit einer Temperaturerhöhung entgegengewirkt. Das Gleichgewicht verschiebt sich in Richtung des Produkts.
Die qualitative Ableitung lässt sich durch eine Rechnung bestätigen. Es sollen zwei Temperaturen T1 und T2 betrachtet werden. Mit der Gibbs-Helmholtz-Gleichung gilt:
ΔH° − T1ΔS0 = −R T1 ln K1
ΔH° − T2ΔS0 = −R T2 ln K2
ΔH° und ΔS0 können näherungsweise als temperaturunabhängig angesehen werden. Auflösen nach ΔS0 und Gleichsetzen ergibt dann:
R ln K1 + ΔH°/T1 = R ln K2 + ΔH°/T2
R ln K2 − R ln K1 = ΔH°/T1 − ΔH°/T2
ln (K2/K1) = ΔH°/R (1/T1 − 1/T2)
| ln (K2/K1) = | ΔH° | ( | 1 | − | 1 | ) |
| R | T1 | T2 |
Für eine exotherme Reaktion (ΔH° < 0) ergibt eine Temperaturerhöhung (T2 > T1) einen negativen Wert für ln K2/K1, K2/K1 ist also <1 oder K2 < K1; das Gleichgewicht verschiebt sich bei Temperaturerhöhung auf die Seite der Ausgangsstoffe, so wie es das Prinzip vom kleinsten Zwang aussagt. Bei endothermen Reaktionen ergibt sich der umgekehrte Fall. Ein Beispiel ist das Boudouard-Gleichgewicht, das im Hochofenprozess Bedeutung hat:
C(s) + CO2(g) ⇄ 2 CO(g) ΔH° = 172,5 kJ mol−1
Bei der Bildung von Kohlenmonoxid wird Wärme verbraucht; das System weicht einer Temperaturerhöhung durch CO-Bildung aus, das Gleichgewicht verschiebt sich auf die Produktseite.
Allgemein gilt also: Temperaturerhöhung führt bei exothermen Reaktionen zu einer Verschiebung des Gleichgewichts in Richtung der Ausgangsstoffe, bei endothermen Reaktionen in Richtung der Endstoffe.
Bei den als Beispiel genannten Reaktionen ändert sich die Gesamt-Molzahl im Gasraum: Bei der Ammoniakbildung entstehen aus 4 mol Ausgangsstoffe 2 mol Endprodukt. Das System kann einer Druckerhöhung ausweichen, indem sich das Gleichgewicht in Richtung der Seite mit der kleineren Molzahl, also in Richtung des Produkts, verschiebt; hierdurch wird die Molzahl im Gasraum verrringert und damit einer Druckerhöhung entgegengewirkt.
Bei der Bildung von CO verdoppelt sich die Molzahl im Gasraum. Druckerhöhung wird folglich das Boudouard-Gleichgewicht in Richtung der Ausgangsstoffe verschieben. Ändert sich die Gesamt-Molzahl bei einer Reaktion nicht (wie im Gleichgewicht H2 + I2 ⇄ 2 HI), so ist die Gleichgewichtslage vom Gesamtdruck unabhängig.
Allgemein gilt: Bei Reaktionen mit Änderung der Gesamt-Molzahl in einer Phase (zum Beispiel dem Gasraum) verschiebt sich das Gleichgewicht durch Konzentrationserhöhung (in der Gasphase: durch Druckerhöhung) in Richtung der Seite mit der kleineren Gesamt-Molzahl.
Es ist nicht notwendig, sich diese Zusammenhänge mit dem sehr blumigen „Prinzip vom kleinsten Zwang“ zu erklären. Die Druckabhängigkeit bei Änderung der Molzahl im Gasraum erschließt sich auch unmittelbar aus der Gleichgewichtskonstante. Hierzu noch einmal das Haber-Bosch-Gleichgewicht. Kp ist für die Bildung von Ammoniak aus den Elementen definiert als:
| p2(NH3) | = Kp |
| p(N2) p3(H2) |
Wird nun der Gesamtdruck auf zum Beispiel das 10-fache erhöht, so wächst der Zähler auf das 100-fache des ursprünglichen Wertes, der Nenner aber auf das 10.000-fache. Die Konstante(!) Kp würde dann auf 1/100 ihres Wertes absinken. Dies wird verhindert, indem die Partialdrücke von N2 und H2 kleiner werden und zugleich der Partialdruck von NH3 zunimmt: das Gleichgewicht hat sich in Richtung auf die Seite mit der kleineren Molzahl im Gasraum verschoben.
Das Haber-Bosch-Verfahren ist das grundlegende technische Verfahren, mit dem die Bereitstellung von Stickstoffdünger gelingt. Seine Bedeutung lässt sich an der Schätzung ablesen, dass die Hälfte aller Stickstoffatome in unserem Körper schon einmal einen Haber-Bosch-Druckreaktor von innen „gesehen“ hat. Dem Prozess liegt eine exotherme Reaktion zugrunde:
3 H2 + N2 ⇄ 2 NH3 ΔrH° = −92,4 kJ mol−1
Die Exothermie der Reaktion ist ein Problem: nach dem Prinzip von Le Chatelier verschiebt sich das Gleichgewicht durch Temperaturerniedrigung in Richtung auf das erwünschte Produkt NH3. Bei Raumtemperatur ist die NH3-Konzentration im Gleichgewicht groß, die Geschwindigkeit der NH3-Bildung aber praktisch Null. Eine ausreichende Aktivierung und damit eine ausreichende Reaktionsgeschwindigkeit wird erst bei Temperaturen erreicht, bei denen die NH3-Konzentration im Gleichgewicht nahezu Null ist. Ein Zünden der Reaktionsmischung wie bei den deutlich stabileren Produkten H2O und HCl ist in diesem Fall nicht möglich. Die Ammoniak-Synthese ist damit ein Beispiel, bei dem das technische Problem im Erreichen des Gleichgewichts liegt. Das Problem lässt sich lösen, indem die erwünschte niedrige Reaktionstemperatur durch eine Erniedrigung der Aktivierungsenergie erzielt wird, also durch Katalyse. Die heute eingesetzten Katalysatoren (oberflächenreiches Eisen mit einigen Prozent Kalium-, Calcium- und Aluminiumoxid-Anteil) beschleunigen die Reaktion so weit, dass bereits ab ca. 400 °C eine merkliche NH3-Bildung einsetzt; in der Technik wird der Temperaturbereich von 400–500 °C verwendet. Bei dieser Temperatur und Normaldruck ist die Ammoniak-Konzentration im Gleichgewicht ca. 0,1 %.
Die bei Normaldruck erzielte Ammoniakmenge ist für eine wirtschaftliche Herstellung von NH3 zu gering. Das MWG zeigt jedoch, dass ein zweiter Parameter nutzbar ist: der Gesamtdruck, dessen Einfluss oben bereits diskutiert wurde. In der Technik wird mit einem Druck von 250–350 bar gearbeitet. Die NH3-Menge im Gleichgewicht beträgt dann 17–18 %, bei der technisch eingesetzten Kontaktzeit zwischen Gas und Katalysator von weniger als 1 min wird ca. 11 % erreicht. Mit einer typischen Menge von ca. 100 Tonnen dieses Katalysators in einem modernen Druckreaktor lassen sich täglich ca. 1.500 Tonnen Ammoniak herstellen.
Der zweite Schritt der technischen Stickstofffixierung ist die Verbrennung des zuerst gebildeten Ammoniaks zu NO. Die Probleme bei der technischen Realisierung dieser Reaktion stellen das Gegenstück zum Haber-Bosch-Verfahren dar. Diente Katalyse dort zur Einstellung des Gleichgewichts, so wird Katalyse beim Ostwald-Verfahren genutzt, um auf keinen Fall das chemische Gleichgewicht zu erreichen. Mit der Reaktion des Ostwald-Prozesses:
4 NH3 + 5 O2 → 4 NO + 6 H2O ΔrH° = −904,6 kJ mol−1
ist nämlich noch keineswegs das Gleichgewicht erreicht. Vielmehr findet das gebildete metastabile NO in der heißen Flamme genug Aktivierungsenergie vor, um gemäß
2 NO → N2 + O2 ΔrH° = −180,8 kJ mol−1
weiter zu reagieren. Die einfache, stark exotherme Verbrennung von Ammoniak führt daher nicht zur Bildung von NO, sondern es entstehen lediglich die stabilen Endprodukte Stickstoff und Wasser.
Versuch 23-13: Verbrennung von Ammoniak über einer NH3-Lösung
Versuch 23-14: Verbrennung von Ammoniak mit Sauerstoff im Daniellhahn
Es fragt sich nach dieser Reaktion natürlich, ob die Formulierung von NO als Zwischenprodukt überhaupt sinnvoll ist. Diese Frage wird vor dem Hintergrund diskutiert, dass sowohl NH3 als auch NO N1-Verbindungen sind.
Der Versuch, die NO-Bildung durch Katalyse zu erreichen, nutzt die bisher zusammengetragenen Erkenntnisse. Beim Ostwald-Verfahren wird gerade verhindert, dass sich das Gasgemisch erhitzt, sondern für die Reaktion wird lediglich eine heiße Katalysatoroberfläche zur Verfügung gestellt:
Versuch 23-24: Katalytische Verbrennung von Ammoniak
Im technischen Verfahren ist die Ausführung weiter optimiert, indem das NH3/Luft-Gemisch in schnellem Strom durch ein Katalysatornetz geleitet wird, wodurch eine Aufenthaltdauer des NO in der heißen Umgebung auf den Bruchteil einer Sekunde verkürzt wird. Es resultiert ein Verfahren, mit dessen Hilfe eine Reaktionsführung fern von Gleichgewichtsbedingungen ermöglicht wird.
Eine Frage zum Schluss: Die zum Nachweis von gebildetem NO dienende Umsetzung
2 NO + O2 → 2 NO2
sollte wegen der oben genannten Werte für die Standardbildungsenthalpien von NO und NO2 exotherm verlaufen. Bei allen Beispielen für exotherme Gasreaktionen mussten diese gezündet werden. Es ist daher überraschend, dass NO selbst bei Raumtemperatur sofort NO2 bildet, wenn es in Kontakt mit Luftsauerstoff kommt. Woran könnte das liegen?
Das Massenwirkungsgesetzes eignet sich zur Beschreibung homogener Gleichgewichte. Eine wichtige Anwendung des MWG ergibt sich jedoch auch für heterogene Gleichgewichte, wenn diese mit homogenen Gleichgewichten vernetzt sind. Ein Beispiel ist die quantitative Beschreibung gesättigter Salzlösungen. Die Löslichkeit eines Stoffes in einem Lösemittel ist die maximale Menge dieses Stoffes, die sich bei einer gegebenen Temperatur in einer bestimmten Menge Lösemittel löst. Eine Lösung, die die größtmögliche Menge an Gelöstem enthält, heißt gesättigte Lösung.
Steht eine gesättigte Lösung mit festem Bodenkörper des gelösten Stoffes im Gleichgewicht, so können – bei einem Salz der Zusammensetzung AB als Beispiel – die folgenden Gleichgewichte formuliert werden:
ABBodenkörper ⇄ ABgelöst ⇄ A+gelöst + B−gelöst
Das linke Gleichgewicht ist ein heterogenes Gleichgewicht. Das rechte, homogene Gleichgewicht beschreibt die im vorigen Kapitel eingeführte Dissoziation eines Salzes in Wasser. Auf dieses homogene Gleichgewicht kann das MWG angewendet werden:
| [A+gelöst] [B−gelöst] | = Kc |
| [ABgelöst] |
[AB] ist das Vielfache einer Einheitskonzentration an gelöstem, undissoziiertem(!) AB. Diese Konzentration, die bei den meisten Elektrolyten unmessbar klein ist, darf als konstant angesehen werden, solange Bodenkörper vorhanden ist, der mit dieser Konzentration über das linke Gleichgewicht verbunden ist. Aus diesem Grund wird [AB] mit der Gleichgewichtskonstante zu einer neuen Konstanten kombiniert, dem Löslichkeitsprodukt LAB:
[A+][B−] = LAB
Für eine Ionenverbindung AB ist also das Produkt der molaren Konzentrationen der (solvatisierten) Ionen in einer gesättigten Lösung für eine gegebene Temperatur eine Konstante. Man beachte, dass Bodenkörper vorhanden sein muss, um ein homogenes Gleichgewicht mit konstantem [AB] formulieren zu können, es ist aber unerheblich, wieviel Bodenkörper vorhanden ist. Da L-Werte etliche Zehnerpotenzen überstreichen, ist es bequem zu definieren (man beachte auch hier, dass nur die entsprechenden dimensionslosen Werte logarithmiert werden):
pL = −lg L
Der Nutzen, ein Löslichkeitsprodukt anstelle einer molaren oder massebezogenen Löslichkeit anzugeben, wird aus dem folgenden Beispiel klar, bei dem das recht schwerlösliche Salz Silberchlorid, AgCl, betrachtet wird. Dieses ist aus Ag+ und Cl−-Ionen aufgebaut. Die Struktur ist dieselbe wie die von Natriumchlorid, allerdings ist AgCl erheblich schwerer löslich als NaCl. Das Löslichkeitsprodukt von Silberchlorid beträgt nur pL(AgCl) = 10. Wegen
AgCl ⇄ Ag+ + Cl−
gilt, dass eine Ausgangskonzentration c0(AgCl) von x mol L−1 in gleiche Mengen Ag+ und Cl− dissoziiert, dass also x mol L−1 Ag+ und x mol L−1 Cl− vorliegen. Es gilt also:
[Ag+] = [Cl−] = c0(AgCl)/mol L−1
Mit pL = 10 ist L = 10−10 und weiter:
L = [Ag+][Cl−] = c02(AgCl)/mol2 L−2 = 10−10
Die molare Löslichkeit ist danach √L · 1 mol L−1, also 10−5 mol L−1.
Das Löslichkeitsprodukt scheint im Vergleich zur molaren Löslichkeit, die sich außerdem leicht in die noch anschaulichere Löslichkeit in g L−1 umrechnen lässt, eine unnötig komplizierte Größe zu sein, um Löslichkeit quantitativ zu beschreiben. Mit dem Löslichkeitsprodukt gelingen nun jedoch auch Berechnungen, bei denen die jeweilige Konzentration einer einzelnen Ionensorte unabhängig vom Gegenion betrachtet wird.
Versuch neu: Gleichioniger Zusatz (NaCl) zu gesättigter AgCl-Lösung
Im Versuch werden der gesättigten AgCl-Lösung Cl−-Ionen zugefügt. Die zugegebene Menge soll zu [Cl−] = 10−2 führen. Wegen
[Ag+] · [Cl−] = [Ag+] · 10−2 = 10−10
gilt:
[Ag+] = 10−10/10−2 = 10−8
Die Konzentration an Ag+-Ionen sinkt also auf 10−8 mol L−1, indem aus jedem Liter der Lösung (10−5 − 10−8) mol ≈ 10−5 mol AgCl ausfällt.
Ein gleichioniger Zusatz setzt also die Löslichkeit eines Salzes herab. Das Gegenteil wird beobachtet, wenn die Sättigung einer Lösung aufgehoben wird. Die einfachste Art ist die Zugabe von weiterem Lösungsmittel, also Verdünnen. Eine Möglichkeit, die Konzentration nur einer Ionensorte zu beeinflussen, besteht auch hier, nämlich die Zugabe eines Reagenzes, das nur mit einem bestimmten Ion eine neue, stabile Lösungsspezies bildet. Ein Beispiel ist die Komplexierung von Ag+ durch fest bindende „Liganden“ wie Ammoniak oder Thiosulfat, S2O32−. Aus dem Alltag bekannt ist die Erhöhung der Löslichkeit von AgBr durch Thiosulfat beim „Fixieren“ photographischer Schichten, bei dem überschüssiges, durch die Entwicklung nicht in Silber überführtes Silberbromid aus der Schicht herausgelöst wird. Der Vorgang zeigt, was mit „[Ag+]“ gemeint ist, nämlich nicht die Konzentration von gelöstem Silber in irgendeiner Form, sondern die Konzentration an gelöstem Silber, das in derselben Form vorliegt, die es auch bei Standardbedingungen einnimmt – bei Kationen also in der Regel das hydratisierte Ion.
Für Elektrolyte anderer Zusammensetzungen als AB ergeben sich für das Löslichkeitsprodukt entsprechende Ausdrücke. Für ein gemäß AB2 ⇄ A2+ + 2 B− dissoziierendes AB2-Salz gilt:
L(AB2) = [A2+][B−]2
Man beachte das bei der Formulierung des MWG übliche Einsetzen der Koeffizienten der Gleichung als Exponenten.
Ein Rechenbeispiel an einer etwas besser löslichen Ionenverbindung aus dem Alltag zeigt noch einmal die Wirkung eines gleichionigen Zusatzes in weniger abstrakter Form. Gegeben ist das Löslichkeitsprodukt von Calciumsulfat, CaSO4 (Gips), von L = 2,2 × 10−4. Die Löslichkeit des AB-Salzes Gips in reinem Wasser errechnet sich damit wie folgt: wegen
L = [Ca2+][SO42−] = c02/mol2 L−2
gilt:
c0 = (2,2 × 10−4)½ = 1,48 × 10−2 mol L−1
Multipliziert mit der Molmasse von 136,14 g mol−1 ergibt sich die Löslichkeit von Gips in Wasser zu 2 g L−1.
Die Löslichkeit von Gips ist kleiner, wenn als Lösungsmittel nicht Wasser, sondern eine Na2SO4-Lösung der Konzentration c = 0,1 mol L−1 verwendet wird. Vernachlässigt man die zusätzliche Sulfatmenge durch die Dissoziation des Gipses, gilt:
[SO42−] = 0,1
Die Konzentration an Ca2+-Ionen sinkt auf
| [Ca2+] = | 2,2 × 10−4 | = 2,2 × 10−3 |
| [SO42−] |
In der Lösung befindet sich also nur noch eine Gesamtmenge von 2,2 × 10−3 mol L−1 CaSO4. Nach Multiplikation mit der Molmasse ergibt sich 0,3 g L−1. Der gleichionige Zusatz hat also die Löslichkeit von Gips von 2 g L−1 auf nur noch 0,3 g L−1 gesenkt.
Der Versuch zum Ausgleich des Elektronenmangels von Verbindungen und Ionen führt zu zwei Grundtypen chemischer Reaktionen: Säure-Base-Reaktionen und Redox-Reaktionen. Während den Elektronenübergängen, den Redox-Reaktionen, das folgende Kapitel gewidmet ist, befasst sich dieses Kapitel mit der „sozialistischen“ Alternative zum Elektronenübergang, nämlich der gemeinsamen Benutzung von Elektronenpaaren zwischen einem elektronenarmen und einem elektronenreichen Reaktionspartner. Alle Säure-Base-Theorien haben die Gemeinsamkeit, das Prinzip „Säure“ mit einem Mangel an negativer oder einem Überschuss an positiver Ladung zu verbinden, während die Base den elektronenreicheren Partner darstellt.
| Mortimer: 18, 19 Binnewies: 10 Riedel: 3.7 |
Ionenverbindungen lösen sich unter Dissoziation in freibewegliche, hydratisierte Ionen in Wasser. Die Lösungen sind elektrisch leitend, Salze sind Elektrolyte. Gase scheinen keinen Bezug zu dieser Chemie zu haben. Liegen jedoch polare kovalente Bindungen vor, so kann es im Sinne einer chemischen Reaktion auch hier zur Ionenbildung kommen.
Versuch 15-8: HCl-Springbrunnen
Versuch 15-9: HCl löst sich exotherm in Wasser
Versuch 15-10: HCl-Lösung leitet den Strom
Versuch 17-1: Reine Essigsäure leitet den Strom nicht, Essigsäurezugabe zu Wasser ergibt eine leitende Lösung
Zur Deutung der extremen Reaktivität von HCl und ähnlicher Stoffe mit Wasser sind im Lauf der Zeit verschiedene Theorien entstanden. Wir werfen nur einen kurzen Blick auf eine der älteren Vorstellungen, der Arrhenius-Theorie, um dann die Theorie kennenzulernen, durch die Säure-Base-Reaktionen vor allem im wässrigen Medium erfolgreich beschrieben werden, der Brønsted-Theorie.
Nach Arrhenius sind Säuren Wasserstoffverbindungen, die in wässriger Lösung unter Bildung von H+-Ionen dissoziieren. Mit HCl als Beispiel ergibt sich:
HCl → H+ + Cl−
Das Gegenstück, Basen, sind Hydroxide, die in wässriger Lösung unter Bildung von OH−-Ionen dissoziieren. Ein Beispiel ist Natriumhydroxid, NaOH:
NaOH → Na+ + OH−
Die Reaktion einer Säure mit einer Base heißt Neutralisation. Aus H+ und OH−-Ionen entsteht Wasser:
H+ + OH− → H2O ΔrH° = −57,4 kJ mol−1
Das Säurerest-Anion und das Kation der Base bleiben bei der Neutralisation unverändert:
Na+ + OH− + H+ + Cl− → Na+ + Cl− + H2O
Die Lösung reagiert nach der Neutralisation weder sauer noch basisch, sondern verhält sich wie eine Lösung von Kochsalz, NaCl, in Wasser. Allgemein gilt für die Neutralisation nach Arrhenius:
Säure + Base → Salz + Wasser
Die Arrhenius-Theorie beschreibt adäquat die exotherme Neutralisation in wässriger Lösung, und sie erklärt den Elektrolytcharakter von Säure- und Base-Lösungen. Unterschiede in der Leitfähigkeit zum Beispiel beim Auflösen verschiedener Säuren wie HCl und Essigsäure können erklärt werden, indem die Dissoziation als Gleichgewichtsreaktion verstanden wird. Säuren, die weitgehend vollständig dissoziieren, sind starke Säuren, während das Dissoziationsgleichgewicht bei einer schwachen Säure wie Essigsäure auf der Seite der undissoziierten Form liegt.
Versuch neu(?): HCl und NH3 bilden NH4Cl
Brønsted erweiterte die Arrhenius-Theorie – vor allem auf der Seite der Base – und betont in seinem Konzept den Charakter von Säure-Base-Reaktionen als typische Gleichgewichtsreaktionen. Brønsted-Säuren sind Protonendonatoren, also Teilchen, die Protonen (H+-Ionen) abspalten können; Basen sind Protonenakzeptoren, das sind Teilchen, die Protonen aufnehmen können. Als Beispiel dient wieder die Reaktion von HCl mit Wasser:
HCl + H2O ⇄ Cl− + H3O+
HCl reagiert als Säure, da ein Proton abgespalten wird, Wasser reagiert als Base, da dieses Proton aufgenommen wird. Im Sinne einer Gleichgewichtsreaktion ist bei der Rückreaktion H3O+ eine Säure, die ein Proton auf das dabei als Base fungierende Cl−-Ion überträgt.
Eine Säure und die durch Protonenabspaltung entstehende Base, im Beispiel also HCl/Cl− und H3O+/H2O, bilden ein konjugiertes Säure-Base-Paar (in der Literatur manchmal: „korrespondierendes“ Paar). Cl− ist die zu HCl konjugierte Base, HCl ist die zu Cl− konjugierte Säure. Allgemein gilt für ein konjugiertes Säure-Base-Paar:
Säure ⇄ Base + H+
Eine Protonenabspaltung von einer Säure allein ist nicht möglich, sondern das im allgemeinen nicht isoliert auftretende Proton wird bei einer Säure-Base-Reaktion von der Säure auf die Base übertragen – es findet eine Protolyse statt. Es lassen sich zwei Teilgleichungen aufstellen, deren Summe den ablaufenden Vorgang beschreibt:
HCl ⇄ Cl− + H+ (Säure-Base-Paar 1)
H2O + H+ ⇄ H3O+ (Säure-Base-Paar 2)
Die Summe der beiden Vorgänge stellt die oben formulierte chemische Reaktion dar, die nach dem Schema abläuft:
Säure 1 + Base 2 ⇄ Base 1 + Säure 2
An einer Protolyse („Brønsted-Säure-Base-Reaktion“) sind also immer zwei Säure-Base-Paare beteiligt. Protolysen führen zu einem Gleichgewicht. Beispiele mit Wasser als Base sind die folgenden Protolysereaktionen:
| Säure 1 | Base 2 | Säure 2 | Base 1 | |||
|---|---|---|---|---|---|---|
| HCl | + | H2O | ⇄ | H3O+ | + | Cl− |
| H2SO4 | + | H2O | ⇄ | H3O+ | + | HSO4− |
| HSO4− | + | H2O | ⇄ | H3O+ | + | SO42− |
| NH4+ | + | H2O | ⇄ | H3O+ | + | NH3 |
| H2O | + | H2O | ⇄ | H3O+ | + | OH− |
| HCO3− | + | H2O | ⇄ | H3O+ | + | CO32− |
Wird, wie im Beispiel, die Reaktion von Säuren in wässriger Lösung betrachtet, so ist eines der beiden Säure-Base-Paare das Paar H3O+/H2O.
Ist wie bei HCl die Tendenz zur Protonenübertragung auf Wasser groß, wenn also das Protolysegleichgewicht auf der rechten Seite liegt, so ist umgekehrt die Tendenz der konjugierten Base zur Protonenaufnahme gering. Eine schwache Säure wie HCO3− hingegen überträgt nur zu einem kleinen Teil Protonen auf Wasser, das Protolysegleichgewicht liegt auf der linken Seite; die konjugierte Base, hier CO32−, ist dementsprechend stark, das heißt, ihre Tendenz zur Aufnahme von Protonen ist groß.
Manche Verbindungen oder Ionen, Ampholyte, können sowohl Protonen aufnehmen als auch abgeben, je nachdem, ob ein stärkerer Protonendonator oder Protonenakzeptor anwesend ist. So kann das HSO4−-Ion von einer Säure protoniert werden, es entsteht die zu HSO4− konjugierte Säure H2SO4; oder HSO4− kann ein Proton an eine Base abgeben, es entsteht die zu HSO4− konjugierte Base SO42−. Wasser kann unter Bildung der konjugierten Säure H3O+, dem Oxonium-Ion, Protonen aufnehmen, es kann aber auch selbst Basen protonieren, wobei das Hydroxid-Ion als konjugierte Base OH− entsteht.
In Abhängigkeit von der Ladung des betrachteten Teilchens wird bisweilen eine Unterteilung von Brønsted-Säuren und -Basen vorgenommen. Positiv geladene Teilchen werden als Kationsäuren oder -basen bezeichnet (Beispiele sind H3O+ und NH4+), ungeladene Säuren und Basen sind Neutralsäuren und -basen, negativ geladene Säuren und Basen schließlich sind Anionsäuren und -basen (Beispiele sind OH−, CO32−, S2−, CN−).
Man beachte, dass nach der Definition von Arrhenius das frei bewegliche H+-Ion Träger der sauren Eigenschaft ist, während nach Brønsted H+ nicht als frei bewegliches Ion formuliert wird, sondern es ist das übertragene Teilchen. Größer ist der Unterschied bei den Basen: nach Arrhenius ist ein Metallhydroxid wie NaOH eine Base, nach Brønsted ist OH− eine von vielen möglichen Basen.
Bei der Protolyse einer Säure in Wasser entstehen nach Arrhenius H+-Ionen, nach Brønsted wird die gleiche molare Menge H3O+ gebildet. Der Säuregrad einer wässrigen Lösung, ihre Acidität, ist durch die Konzentration an H+ bzw. H3O+ gegeben. Die in der Praxis wichtigen Konzentrationsangaben überstreichen mehrere Zehnerpotenzen, so dass die Einführung eines logarithmischen Maßes zu handlicheren Zahlenwerten führt. Es wird daher ein pH-Wert als negativer dekadischer Logarithmus der H+- bzw. der H3O+-Konzentration definiert (im folgenden wird H3O+ formuliert, die Aussagen dieses Abschnittes lassen sich aber auch mit der Arrheniusschen Vorstellung entwickeln):
pH = −lg [H3O+]
In reinem Wasser lässt sich ein nahezu vollständig auf der linken Seite liegendes Protolysegleichgewicht nachweisen, in dem H2O sowohl als Säure wie auch als Base vorkommt; Wasser zeigt sich als Ampholyt, der Autoprotolyse zeigt (nach Arrhenius: Eigendissoziation):
H2O + H2O ⇄ H3O+ + OH−
Mit dem MWG wird erhalten:
| [H3O+] · [OH−] | = K |
| [H2O]2 |
Da nur wenige H2O-Moleküle Ionen bilden, ist die Konzentration des nicht protolysierten Wassers praktisch gleich der Gesamtkonzentration an Wasser:
c0(H2O) = 1000 g L−1/18 g mol−1 = 55,5 mol L−1
Diese praktisch konstante Konzentration an H2O wird mit der Gleichgewichtskonstanten zu einer neuen Konstante zusammengefasst:
[H3O+] · [OH−] = K · [H2O]2 = KW
KW ist das Ionenprodukt des Wassers. Sein Zahlenwert beträgt bei 25 °C:
KW = 1,0 · 10−14
Aufgrund des Autoprotolysegleichgewichtes ist also das Produkt der Konzentrationen der H3O+- und OH−-Ionen in Wasser und auch in verdünnten wässrigen Lösungen konstant. Mit pOH = −lg [OH−] ergibt sich nach dem Logarithmieren:
pH + pOH = 14
Für reines Wasser gilt:
[H3O+] = [OH−] = KW½ = 10−7
und:
pH = 7
Liegt in einer wässrigen Lösung eine hiervon abweichende H3O+- oder OH−-Ionen-Konzentration vor, so ergibt sich die Konzentration der jeweils anderen Ionensorte aus dem Ionenprodukt. So ist zum Beispiel in einer Lösung mit [H3O]+ = 10−2 die OH−-Konzentration:
| [OH]− = | KW | = | 10−14 | = 10−12 |
| [H3O+] | 10−2 |
oder, mit logarithmierten Werten:
pH = 2
und:
pOH = 14 − pH = 14 − 2 = 12
Im Beispiel ist die Konzentration der H3O+-Ionen größer als die Konzentration der OH−-Ionen; die Lösung reagiert sauer. Allgemein sind wässrige Lösungen sauer für pH < 7 und basisch (alkalisch) für pH > 7. In der Praxis reicht die pH-Skala ungefähr von 0 bis 14. Jenseits dieser Grenzen sind die Lösungen so konzentriert, dass die Zahlenangaben mit immer größeren systematischen Fehlern behaftet sind.
Man beachte, dass die Autoprotolyse des Wassers als Umkehrung der exotherm verlaufenden Neutralisation ein endothermer Vorgang ist, dessen Temperaturabhängigkeit nach den im vorigen Kapitel eingeführten Regeln ermittelt werden kann. (Nun?)
Die Lage des Protolysegleichgewichts einer Säure HA mit der Base Wasser,
HA + H2O ⇄ A− + H3O+,
wird dazu benutzt, die Stärke einer Säure quantitativ zu erfassen. Eine starke Säure mit einer großen Tendenz zur Deprotonierung ergibt ein weiter auf der rechten Seite liegendes Gleichgewicht als eine schwache Säure. Bei der Formulierung des MWG gemäß
| [H3O+] · [A−] | = K |
| [HA] · [H2O] |
kann die in verdünnten Lösungen konstante Konzentration von Wasser, [H2O], mit der Massenwirkungskonstante K multipliziert werden:
| [H3O+] · [A−] | = K · [H2O] = KS |
| [HA] |
KS ist die Säurekonstante. Da KS-Werte den Bereich etlicher Zehnerpotenzen überstreichen, wird auch hier ein pKS-Wert als negativer Zehnerlogarithmus des KS-Wertes definiert:
pKS = −lg KS
Bei starken Säuren überwiegt die rechte Seite des Protolysegleichgewichts, der Zähler in der den KS-Wert definierenden Gleichung ist größer als der Nenner; es gilt: KS > 1 und pKS < 0. Je schwächer eine Säure ist, um so größer ist ihr pKS-Wert. Für die im nächsten Abschnitt behandelten pH-Wert-Berechnungen genügt eine Einteilung in zwei Gruppen:
• starke Säuren: pKS < 0
• schwache Säuren: pKS > 0
Mehrprotonige (mehrbasige) Säuren, die wie Schwefelsäure mehrere Protonen abspalten können, besitzen von Stufe zu Stufe eine um ca. 5 pKS-Einheiten abnehmende Säurestärke. Schwefelsäure ist ein Beispiel für eine zweiprotonige Säure (weitere Beispiele: Kohlensäure, H2CO3; schweflige Säure, H2SO3), eine dreiprotonige Säure ist zum Beispiel Phosphorsäure, H3PO4.
Das Protolysegleichgewicht einer starken Säure (pKS < 0) wie HCl oder H2SO4 liegt praktisch vollständig auf der rechten Seite des jeweiligen Protolysegleichgewichts
HCl + H2O ⇄ Cl− + H3O+
H2SO4 + H2O ⇄ HSO4− + H3O+
Praktisch alle Säuremoleküle protonieren H2O zu H3O+, pro HCl- bzw. H2SO4-Molekül entsteht also ein H3O+-Ion. Die H3O+-Konzentration der Lösung ist daher gleich der Gesamtkonzentration c0 der Säure, so dass für den pH-Wert gilt:
pH = −lg c0/mol L−1
So errechnet sich der pH-Wert einer HCl-Lösung der Konzentration 0,1 mol L−1 zu
pH = −lg 0,1 = 1
Der pH-Wert einer HClO4-Lösung der Konzentration 0,5 mol L−1 beträgt:
pH = −lg 0,5 = 0,3
Das Protolysegleichgewicht einer schwachen Säure (pKS > 0) wie Essigsäure, CH3COOH, abgekürzt HOAc (OAc für Acetat, dem Säurerest der Essigsäure [lat. acetum, Essig]) liegt dagegen weitgehend auf der linken Seite:
HOAc + H2O ⇄ OAc− + H3O+
Die Berechnung des pH-Wertes der Lösung ergibt sich aus dem pKS-Wert
| [H3O+] · [OAc−] | = KS |
| [HOAc] |
Da in der Lösung, die neben Wasser nur Essigsäure enthält, als Folge des Protolysegleichgewichts gleiche molare Mengen H3O+ und OAc− vorliegen, gilt:
[OAc−] = [H3O+]
Durch Einsetzen ergibt sich:
| [H3O+] · [OAc−] | = | [H3O+]2 | = KS |
| [HOAc] | [HOAc] |
Wegen der kleinen Säurekonstante, KS(HOAc) ist zum Beispiel 1,8 · 10−5, gilt:
[HOAc] ≈ c0(HOAc)/mol L−1
Die Konzentration an unprotolysierter Säure entspricht also ungefähr der Gesamtkonzentration c0. Für pH ergibt sich mit dieser Näherung, die für pKS > ≈ 3 zu keinem nennenswerten Fehler führt:
[H3O+]2 = [HOAc] · KS = c0/mol L−1 · KS
und weiter:
[H3O+] = (KS · c0/mol L−1)1/2
Logarithmen ergibt:
pH = ½ (pKS − lg c0/mol L−1)
So ist der pH-Wert einer Essigsäurelösung der Ausgangskonzentration c0 = 0,1 mol L−1:
pH = ½ (4,74 − (−1)) = 2,87
Der pH-Wert der Essigsäurelösung ist erwartungsgemäß größer als der einer gleichkonzentrierten Salzsäure- oder Schwefelsäurelösung (pH = 1). Der um etwa 2 pH-Einheiten größere Wert bei der Essigsäure bedeutet eine H3O+-Ionenkonzentration, die etwa das 10−2-fache, also 1/100 von [H3O+] beträgt. Die Essigsäure ist damit bei einer Gesamtkonzentration von 0,1 mol L−1 nur zu etwa 1 % deprotoniert: der Protolysegrad α, der Anteil an protolysierter Säure an der Gesamtmenge, ist nur gering. Diese anschauliche Größe kann auch direkt berechnet werden. Wegen
[H3O+] = α · c0/mol L−1,
[A−] = α · c0/mol L−1, und
[HA] = (1 − α) · c0/mol L−1,
gilt:
| α2 | c0 = KS |
| 1 − α |
Für eine schwache Säure mit α ≈ 0 ist (1 − α) ≈ 1, so dass gilt:
| α = ( | KS | )½ |
| c0/mol L−1 |
Für Essigsäure der Gesamtkonzentration 0,1 mol L−1 und KS ≈ 10−5 ergibt sich dann der oben genannte Wert von 1 %. Mit wachsender Verdünnung steigt der Protolysegrad einer schwachen Säure, um bei unendlicher Verdünnung den Wert für starke Säuren zu erreichen. Man vergleiche zum Beispiel die pH-Werte von Säuren der Konzentration 0,001 mol L−1:
(a) Starke Säuren:
pH = − lg c0 = − lg 10−3 = 3
(b) Essigsäure:
pH = ½ (pKS − lg c0) = ½ (4,74 − (−3)) = 3,9
Bei der größeren Verdünnung von 1/1000 mol L−1 beträgt der Unterschied der pH-Werte nur noch ca. 1 Einheit, [H3O+] beträgt nun bei Essigsäure ca. 0,1 mol L−1, entsprechend einer Protolyse von ca. 10 % der Essigsäuremoleküle. Das Ergebnis ergibt sich auch durch α = (10−5/10−3)½ = 0,1.
Die Berechnungen werden spürbar ungenau, wenn ungünstige Bedingungen vorliegen. So tritt ein merklicher Fehler in zwei nah verwandten Fällen auf, und zwar
• bei mehrprotonigen Säuren (bei denen wir den pH-Wert nur unter Berücksichtigung der ersten Stufe berechnen), wenn c0 ≤ KS2. Wenn dies zutrifft, protolysiert auch die zweite Stufe praktisch vollständig;
• bei schwachen Säuren, wenn c0 ≤ KS. Wenn dies zutrifft, protolysiert die Säure praktisch vollständig.
Mag man sich im Grenzgebiet zwischen starker und schwacher Säure nicht für eine Gleichung entscheiden oder benötigt man auch hier eine präzise Angabe, würde man auf die Näherung
[HA] ≈ c0/mol L−1
verzichten. Die Gleichung
[H3O+]2 = c0/mol L−1 · KS
würde nun so aussehen:
[H3O+]2 = (c0/mol L−1 − [H3O+]) · KS
so dass sich aufgrund des (c0/mol L−1 − [H3O+])-Terms eine quadratische Ergänzung ergeben würde, wodurch die pH-Wert-Berechnung etwas aufwendiger würde.
Hydroxide wie NaOH, KOH oder Ca(OH)2 dissoziieren in Wasser vollständig gemäß
NaOH → Na+ + OH−
Eine NaOH-Lösung der Konzentration 0,1 mol L−1 enthält daher 0,1 mol L−1 OH−-Ionen. Mit pOH = −lg [OH−] und pH + pOH = 14 ergibt sich für den pH-Wert:
[OH−] = 0,1
pOH = − lg 0,1 = 1
pH = 14 − pOH = 14 −1 = 13
Analog ergibt sich für eine Ca(OH)2-Lösung („Löschkalk“-Lösung) der Konzentration 0,001 molL−1 wegen der Bildung von 2 OH−-Ionen aus einer Formeleinheit Ca(OH)2:
[OH−] = 0,002
pOH = − lg 0,002 = 2,7
pH = 14 − pOH = 14 −2,7 = 11,3
Bei der Auflösung von Oxiden in Wasser findet die folgende Protolyse statt:
O2− + H2O → 2 OH−
Ein Beispiel ist das „Löschen“ (Umsetzen mit Wasser) von gebranntem Kalk:
CaO + H2O → Ca(OH)2
Beim Auflösen von 0,001 mol Calciumoxid, CaO, in 1 L Wasser ergibt sich also eine Lösung mit dem gleichen pH-Wert wie bei der Ca(OH)2-Lösung.
Die Reaktion von Branntkalk mit Wasser zeigt ein wichtiges Prinzip: Das Hydroxid-Ion ist die stärkste Base, die in Wasser existieren kann. Kommt eine noch stärkere Base wie das Oxid-Ion mit Wasser in Kontakt, so wird sie sofort unter Bildung von Hydroxid-Ionen protoniert.
Dasselbe gilt für den Fall einer Säure, die stärker sauer ist als das Oxonium-Ion. HCl ist ein Beispiel; mit einem pKS von −6 ist HCl um einige Zehnerpotenzen saurer als das Oxonium-Ion, so dass das Protolyse-Gleichgewicht vollständig auf der Seite des H3O+-Ions liegt. Die praktisch vollständige Umsetzung von Oxid-Ionen und HCl-Molekülen führt zu derselben Hydroxid- und Oxonium-Ionen-Konzentration wie eine Reaktion noch stärkerer Basen und Säuren mit Wasser – Wasser ist ein nivellierendes Lösemittel.
Die im vorigen Abschnitt behandelte Dissoziation von NaOH in Wasser in Na+ und OH−-Ionen führt zu einer Lösung, deren pH-Wert hinreichend genau berechnet werden kann, wenn nur der Einfluss der OH−-Ionen berücksichtigt wird. Die Na+-Ionen liegen in der Lösung hydratisiert vor, zum Beispiel als [Na(H2O)6]+. Hydratisierte Kationen, vor allem Kationen mit einem Metallion höherer Ladung wie [Fe(H2O)6]3+ oder [Al(H2O)6]3+, wirken als Protonen-Donatoren, wenn sie mit Wasser als Base Protolysegleichgewichte der Art
[Al(H2O)6]3+ + H2O ⇄ [Al(H2O)5(OH)]2+ + H3O+
eingehen. Der pH-Wert der Lösung wird hierdurch erniedrigt. Im Fall der Hydroxide mit Na+ und Ca2+ sind die hydratisierten Ionen so schwach sauer, dass sie zum pH-Wert der Lösung keinen Betrag leisten.
Zur pH-Wert-Berechnung wird von der Überlegung ausgegangen, dass Salze wie Aluminiumchlorid, AlCl3, Eisen(III)-chlorid, FeCl3, Ammoniumchlorid, NH4Cl, oder Kaliumhydrogensulfat, KHSO4, wie fast alle Salze in Wasser vollständig und unter Hydratisierung der Ionen dissoziieren:
AlCl3 → Al3+(aq) + 3 Cl−(aq)
FeCl3 → Fe3+(aq) + 3 Cl−(aq)
NH4Cl → NH4+(aq) + Cl−(aq)
KHSO4 → K+(aq) + HSO4−(aq)
Hydratisierte K+- und Cl−-Ionen sind so schwach sauer bzw. basisch, dass deren Protolyse den pH-Wert der Lösung praktisch nicht beeinflusst. Die übrigen Ionen hingegen sind in hydratisierter Form schwache Brønsted-Säuren, die entsprechend ihrem pKS-Wert Wasser zu H3O+ protonieren können:
[Al(H2O)6]3+ + H2O ⇄ [Al(H2O)5(OH)]2+ + H3O+ pKS = 4,9
[Fe(H2O)6]3+ + H2O ⇄ [Fe(H2O)5(OH)]2+ + H3O+ pKS = 2,2
NH4+ + H2O ⇄ NH3 + H3O+ pKS = 9,2
HSO4− + H2O ⇄ SO42− + H3O+ pKS = 1,9
Der pH-Wert der Lösungen errechnet sich auf die gleiche Weise wie bei den oben angeführten schwachen Säuren:
pH = ½ (pKS − lg c0/mol L−1)
Für Lösungen der Konzentration 0,1 mol L−1 ergibt sich jeweils:
AlCl3: pH = ½ (4,9 − (−1)) = 3,0
FeCl3: pH = ½ (2,4 − (−1)) = 1,7
NH4Cl: pH = ½ (9,3 − (−1)) = 5,1
KHSO4: pH = ½ (1,9 − (−1)) = 1,5
Die für FeCl3 und KHSO4 errechneten Werte sind mit einem größeren Fehler behaftet, da die pKS-Werte schon recht nahe am vorgegebenen Wert für c0 liegt, dass die Näherung „schwache Säure“ nicht voll zulässig ist.
Wässrige Lösungen von Natriumacetat oder Natriumcarbonat („Soda“) weisen pH-Werte im basischen Bereich auf. Dies ist auf die folgenden Protolyse-Gleichgewichte zurückzuführen:
OAc− + H2O ⇄ HOAc + OH−
CO32− + H2O ⇄ HCO3− + OH−
Die Basizität dieser Anionbasen wird durch die Basekonstante, den KB-Wert, beschrieben, der analog zum KS-Wert definiert wird; zum Beispiel mit Acetat:
| [OH−] · [HOAc] | = KB |
| [OAc−] |
Allgemein mit einer Base B formuliert, gilt für die Basekonstante:
B + H2O ⇄ HB+ + OH−
| [OH−] · [BH+] | = KB |
| [B] |
Analog gilt weiter:
pKB = −lg KB
pKB-Werte sind nur selten tabelliert, da sie aus dem pKS-Wert der konjugierten Säure leicht errechnet werden können. Für ein konjugiertes Säure-Base-Paar HA/A− gilt nämlich (A− jetzt als B):
| [H3O+] · [A−] | = KS |
| [HA] |
und
| [OH−] · [HA] | = KB |
| [A−] |
Multiplizieren ergibt:
| KS · KB = | [H3O+] · [A−] | · | [OH−] · [HA] | = [H3O+] · [OH−] = KW |
| [HA] | [A−] |
Nach dem Logarithmieren und dem Einsetzen des Zahlenwertes für KW bei Raumtemperatur ergibt sich für ein konjugiertes Säure-Base-Paar:
pKS + pKB = 14
So ergibt sich der pKB-Wert von Acetat zu:
pKB(OAc−) = 14 − pKS(HOAc) = 14 − 4,7 = 9,3
Der weitere Formalismus entspricht völlig dem Vorgehen bei schwachen Säuren (beachte die analoge Unterscheidung: schwache Base mit pKB > 0, starke Base mit pKB < 0); es ergibt sich für pOH:
pOH = ½ (pKB − lg c0/mol L−1)
Zum pH-Wert gelangt man dann durch pH = 14 − pOH. So ist der pH-Wert von Soda-Lösung der Konzentration 0,1 mol L−1:
pKB(CO32−) = 14 − pKS(HCO3−) = 14 − 10,3 = 3,7
Das Carbonat-Ion ist mit diesem pKB-Wert eine schwache Base, so dass gilt:
pOH = ½ (3,7 − (−1)) = 2,4
pH = 14 − 2,4 = 11,6
Die für Anionbasen entwickelten Formeln gelten auch für Neutralbasen wie Ammoniak, NH3. Ein Beispiel ist die Berechnung des pH-Wertes einer wässrigen Ammoniaklösung der Konzentration 0,01 mol L−1:
pKB(NH3) = 14 − pKS(NH4+) = 14 − 9,3 = 4,7
Mit diesem pKB-Wert ist Ammoniak eine schwache Base, so dass gilt:
pOH = ½(4,7 − (−2)) = 3,4
pH = 14 − 3,4 = 10,6
Und wie reagiert eine wässrige Lösung von Ammoniumacetat, NH4+OAc−? Säure und Base finden nun andere Reaktionspartner vor. Das Protolysegleichgewicht des Ammonium-Ions ist neu zu formulieren. In Abwesenheit des Acetats galt:
NH4+ + H2O ⇄ NH3 + H3O+
Das Ammonium-Ion überträgt sein Proton auf Wasser, das mit einem pKB-Wert von 14 − pKS(H3O)+ = 14 − (−1,7) = 15,7 eine schwache Base ist. Nun aber ist mit dem Acetat-Ion eine deutlich stärkere Base anwesend (pKB = 9,3; siehe oben). Vom Acetat-Ion ausgehend hätte die Überlegung dasselbe Ziel gehabt, dass nämlich das einzige maßgebliche Protolyse-Gleichgewicht nun lautet:
NH4+ + OAc− ⇄ NH3 + HOAc
Wird das Salz allgemein als HB+A− formuliert, gilt:
HB+ + A− ⇄ B + HA
Der pH-Wert der Lösung errechnet sich gemäß:
pH = ½ {pKS(BH+) + pKS(HA)}
Auf die Formel wird weiter unten näher eingegangen. Man beachte bereits jetzt, dass im Gegensatz zu allen bisher behandelten Fällen der pH-Wert hier von c0 unabhängig ist.
Ammoniumacetat ist ein Ampholyt, da einem Paar aus schwacher Säure und schwacher Base mit dem jeweiligen Gegenion eine stärkere Base und eine stärkere Säure als Protolysepartner zur Verfügung steht als Wasser. Allgemein ist ein Salz BH+A− immer dann ein Ampholyt, wenn die pKS-Werte von BH+ und HA im Bereich von −1,7 und 15,7 liegen. Moleküle sind dann Ampholyte, wenn sie zwei funktionelle Gruppen mit dieser Eigenschaft tragen. Ein bedeutendes Beispiel sind die von den Aminosäuren in wässriger Lösung gebildeten Zwitterionen, bei denen man sich die protolysierenden Gruppen des Ammoniumacetats miteinander kovalent verbunden vorstellen kann. Ein Beipiel ist Glycin, die einfachste Aminosäure:
Das Protolysegleichgewicht liegt auf der rechten Seite (siehe weiter unten); der pH-Wert der Lösung errechnet sich mit derselben Gleichung wie bei Ammoniumacetat als Mittelwert des pKS-Wertes der Ammoniumfunktion von 9,8 und des pKS-Wertes der zur Carboxylatgruppe konjugierten Säure von 2,4 zu 6,1. Dieser pH-Wert ist der isoelektrische Punkt der Aminosäure. In Lösungen mit höherem pH-Wert liegt Glycin zunehmend als Anion
H2N–CH2–COO−
vor, bei pH-Werten unterhalb des isoelektrischen Punktes zunehmend als Kation
H3N+–CH2–COOH.
Moleküle oder Ionen, bei welchen eine einzige funktionelle Gruppe sowohl als Brønsted-Säure als auch als -Base fungieren kann, sind ebenfalls Ampholyte, wenn die pKS-Werte dieser Spezies und ihrer konjugierten Säure im Bereich zwischen −1,7 und 15,7 liegen. Beispiele sind das Dihydrogenphosphat(1−)-Ion, H2PO4−, und das Hydrogenphosphat(2−)-Ion, HPO42−, und, als Grenzfall, auch Wasser. Um den pH-Wert von Lösungen dieser Ionen zu berechnen, ist die bisher benutzte Formel zu verallgemeinern, so dass auch dieser neue Typ mit eingeschlossen ist. Hierzu wird der Ampholyt als X bezeichnet, seine konjugierte Base als B und seine konjugierte Säure als S. X ist Teil der folgenden drei Protolysegleichgewichte:
X + H2O ⇄ B + H3O+
X + H2O ⇄ S + OH−
X + X ⇄ S + B
Während die untere Gleichung das dominierende Protolysegleichgewicht beschreibt, enthalten die oberen beiden Gleichungen die für die pH-Wert-Berechnung maßgebliche Information. Nun werden die Gleichungen für KS(X) und KS(S) aufgestellt. Nach Multiplikation der beiden KS-Werte, Vereinfachung durch die Randbedingung [S] = [B] aufgrund des dominierenden Protolysegleichgewichts, Auflösen der Gleichung nach [H3O+] und Bildung des negativen Logarithmus wird erhalten:
pH = ½ {pKS(X) + pKS(S)}
Mit den pKS-Werten der drei Protolysestufen der Phosphorsäure von 2,1 sowie 7,2 und 12,0 ergeben sich die pH-Werte von Lösungen des Dihydrogenphosphats:
pH = ½ (2,1 + 7,2) = 4,7
und des Monohydrogenphosphats:
pH = ½ (7,2 + 12,0) = 9,6
Mit den Werten für pKS(X) und pKS(S) lässt sich auch die Lage des wesentlichen Gleichgewichts
X + X ⇄ S + B
ermitteln. Formulieren Sie hierzu die Massenwirkungskonstante K dieses Gleichgewichts und drücken Sie die beiden Quotienten aus Säure und jeweiliger konjugierter Base durch die beiden Konstanten KS(X) und KS(S) aus. Es ergibt sich:
K = KS(X) · KS(S)−1
Für eine Lösung von Dihydrogenphosphat gilt wegen KS(X) = 10−7,2 und KS(S) = 10−2,1:
K = 10−7,2/10−2,1 = 10−5,1
Die Lösung enthält also kaum Phosphorsäure und Hydrogenphosphat, sondern praktisch ausschließlich den Ampholyten Dihydrogenphosphat.
Dieselbe Gleichung kann auch auf Ammoniumacetat oder auf Aminosäuren am isoelektrischen Punkt angewendet werden. KS(S) ist hier die Säurekonstante der Säure, die durch Protonierung des Ampholyten erhalten wird (Essigsäure oder die Carboxylfunktion der Aminosäure), KS(X) ist die Säurekonstante der schwächeren Säure im Gleichgewicht (Ammoniumion oder Ammoniumfunktion der Aminosäure). Für das oben formulierte Gleichgewicht
NH4+ + OAc− ⇄ NH3 + HOAc
ist daher zuzuordnen:
2 X ⇄ B + S
Mit KS(X) = KS(NH4+) = 10−9,3 und KS(S) = KS(HOAc) = 10−4,7 gilt also:
K = KS(X) · KS(S)−1 = 10−9,3/10−4,7 = 10−4,6
Analog ergibt sich für Glycin:
K = KS(X) · KS(S)−1 = 10−9,8/10−2,4 = 10−7,4
Das Glycin-Zwitterion liegt also in einer Glycin-Lösung am isoelektrischen Punkt in mehr als der 107-fachen Menge gegenüber der nichtionischen Amino-Carboxyl-Form (B + S) vor.
Versuch neu: Phosphatpuffer • die Zugabe kleiner Mengen Salzsäure und Natronlauge zu Wasser führt zu drastischen pH-Wert-Sprüngen. Diese bleiben jedoch aus, wenn das reine Wasser durch einen NaH2PO4/Na2HPO4-Puffer ersetzt wird. Auch größere HCl- und NaOH-Mengen bewirken im Puffer keine durch die Indikatoren Phenolphthalein und Methylrot angezeigten pH-Sprünge.
In der belebten Natur und in der Laborpraxis ist es oft wichtig, größere pH-Wert-Verschiebungen zu verhindern. Entsteht als Beispiel in einer Zelle durch die Wirkung eines esterspaltenden Enzyms aus einem Ester Alkohol und freie Säure, so würde die Säurefreisetzung zu einem für die Zelle schädlichen Absinken des pH-Wertes führen. Dies wird durch Puffergemische verhindert, bei dem eine schwache Säure im Gemisch mit ihrer ebenfalls schwachen konjugierten Base vorliegt. Ein Beispiel aus der biochemischen Laborpraxis ist der NaH2PO4/Na2HPO4-Puffer. Wird einer solchen Lösung aus diesen beiden Salzen eine starke Säure zugesetzt, so wird diese die basische Komponente des Puffers protonieren, also eine äquivalente Menge an HPO42−. Dabei entsteht die konjugierte Säure der basischen Komponente, die bei einem Puffer eine schwache Säure ist – hier das H2PO4−-Ion. In der Bilanz ist die zugesetzte Menge an starker Säure in die äquivalente Menge einer schwachen Säure umgewandelt worden. Damit zeigt die Pufferlösung als Folge der Zugabe von starker Säure eine kleinere pH-Wert-Änderung als ein gleiches Volumen reinen Wassers.
Weitere wichtige Puffergemische sind:
HOAc + NaOAc
NH4+-Salze + NH3
(H2CO3) + HCO3−
Das letztgenannte Puffergemisch ist von besonderer Bedeutung; es ist eine wichtige Komponente des Puffersystems des Blutplasmas, mit dessen Hilfe der physiologische pH-Wert (7,36 beim Menschen) aufrecht erhalten wird. Die Formel der Kohlensäure ist in Klammern formuliert; dies soll andeuten, dass Kohlensäure selbst wieder im Gleichgewicht mit physikalisch gelöstem CO2 steht.
Der pH-Wert einer Pufferlösung errechnet sich für jedes Mischungsverhältnis von HA und A− nach der Puffergleichung, die auch als Henderson-Hasselbalch-Gleichung bekannt ist. Sie entsteht durch bloßes Umformen und Logarithmieren der Definitionsgleichung der Säurekonstanten. So folgt aus
| [H3O+] · [A−] | = KS |
| [HA] |
durch Umstellen
| [H3O+] = KS · | [HA] |
| [A−] |
Logarithmieren, Multiplikation mit (−1) und Einsetzen von pH und pKS ergibt dann:
| pH = pKS − lg | [HA] |
| [A−] |
Man beachte, dass der pH-Wert eines Puffers aus gleichen molaren Anteilen Säure und Base gerade dem pKS-Wert der Säure entspricht. Die Säurekonstante der sauren Komponente legt damit den pH-Wert fest, um den herum der Puffer wirksam ist.
Die graphische Auftragung der Puffergleichung ergibt eine flach verlaufende Pufferungskurve, die bei der 1:1-Zusammensetzung einen Wendepunkt aufweist, den Pufferpunkt. Im folgenden Bild ist eine solche Kurve für den pKS-Wert 7,0 aufgestellt. Die rot dargestellte Kurve hängt nicht von der Gesamtkonzentration der Komponenten HA und A− ab, sondern nur von deren Molverhältnis. Wichtig wird die Gesamtkonzentration erst, wenn nach der (hier nicht in Zahlen behandelten) Pufferkapazität gefragt wird. Das Bild zeigt die Situation für eine Gesamtkonzentration von 0,5 mol L−1 sowohl an Säure wie auch an Base. Bei Zusatz starker Säure reagiert diese mit der Base des Puffers, bis ein Zusatz von 0,5 mol L−1 Säure alle Base verbraucht hat; der linke Rand der Pufferungskurve ist nun erreicht, es liegt nur noch HA vor. In gleicher Weise führt der Zusatz starker Base zur Reaktion mit HA und dessen schließlich vollständiger Umwandlung in A− am rechten Rand der Pufferungskurve. Man vergleiche die geringe Veränderung des pH-Wertes vor allem um den Pufferpunkt herum mit dem Vergleichsexperiment, bei dem starke Säure oder Base zu reinem Wasser gegeben wird (graue Kurve in der Abbildung).
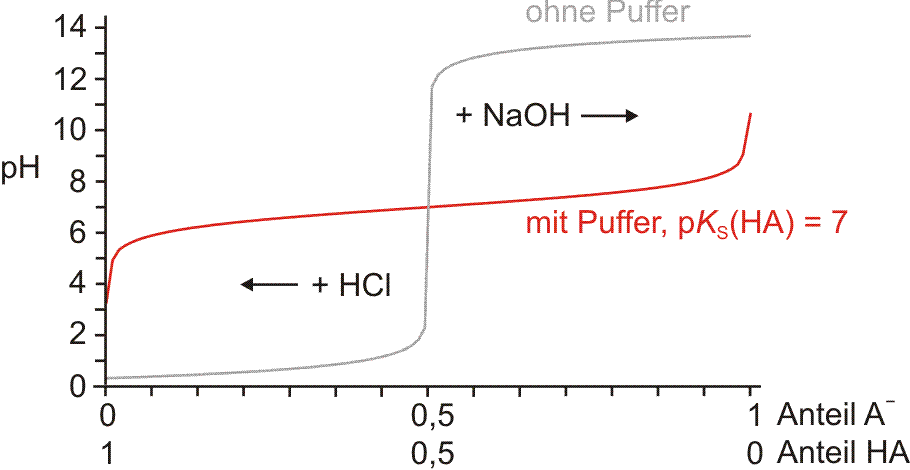
Pufferungskurven sind Teilmengen von Titrationskurven schwacher Säuren mit einer starken Base oder schwacher Basen mit starker Säure. Eine Titrationskurve entsteht, wenn zum Beispiel einer schwachen Säure schrittweise starke Base zugefügt wird:
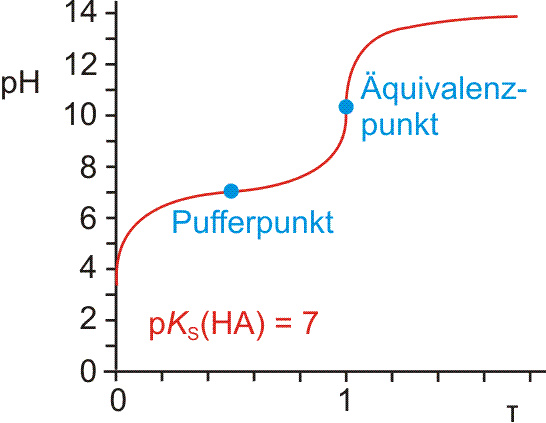
Titration einer schwachen Säure mit einer starken Base. Auf der Abszisse ist der Titrationsgrad τ abgetragen; τ = 1 entspricht dem Zusatz einer äquivalenten Menge an Titrierlösung. Die Auftragung geht von einer konstanten Gesamtkonzentration von 0,5 mol L−1 aus. Dies führt zu den pH-Werten 3,7 sowie 10,3 und 13,7 für τ = 0, 1 und >>1 (wie?).
Neben dem Pufferungspunkt beim pKS-Wert der Säure weist die Kurve im Anschluss an den Pufferbereich den Äquivalenzpunkt auf, bei dem die Säure mit der äquivalenten Menge Base umgesetzt ist. Am Äquivalenzpunkt liegt A− vor und bestimmt den pH-Wert der Lösung (nämlich?).
Man beachte, dass die Aufnahme einer Titrationskurve und die Identifizierung des Pufferpunktes das Standardverfahren darstellt, um den pKS-Wert einer Säure experimentell zu bestimmen.
Versuch 17-7 (nur Blaukrautteil): Blaukrautsaft als Universalindikator
Indikatoren sind synthetische (Methylrot, Phenolphthalein) oder auch natürliche (Lackmus aus Flechten, Blaukrautsaft) Farbstoffe, die in einem charakteristischen pH-Bereich, dem Umschlagsbereich, ihre Farbe verändern. Ein Farbstoff kann als Indikator genutzt werden, wenn er eine schwache Säure oder Base darstellt, deren konjugierte Formen eine andere Farbe aufweisen. Beide Formen liegen im folgenden pH-abhängigen Protolyse-Gleichgewicht vor, das quantitativ durch die Puffergleichung beschrieben wird:
HInd + H2O ⇄ Ind− + H3O+
Am Umschlagspunkt liegt ein 1:1-Gemisch der beiden Formen HInd und Ind− vor; es gilt: [HInd] = [Ind−]. Nach der Puffergleichung ist der pH-Wert am Umschlagspunkt gleich dem pKS-Wert der Indikatorsäure HInd. Wie auch bei farblosen Puffern gilt für die Gemische aus HInd und Ind−, dass in einem pH-Bereich von ±1 um den Umschlagspunkt das Verhältnis [HA]/[A−], hier [HInd]/ [Ind−], zwischen 10 und 0,1 variiert. Da bei einem jeweils zehnfachen Überschuss einer der beiden farbigen Teilchen das Auge nur die im Überschuss vorliegende Farbe wahrnimmt, ist für den Umschlagsbereich eines Indikators meist ein pH-Bereich von ≈2 angegeben.
Geschickt gewählte Mischungen von Indikatoren dienen als „Universalindikatoren“ zur pH-Bestimmung in wässriger Lösung. Bei diesen Indikatormischungen ist meist über den pH-Bereich von 0 bis 14 jedem pH-Wert in Intervallen von 0,5 bis 1 eine Farbe zugeordnet.
Die schnelle und genaue pH-Wert-Messung sowie die kontinuierliche pH-Überwachung gelingt auf elektrochemischem Wege durch pH-Meter. Diese galvanischen Elemente bedienen sich der Nernstschen Gleichung, nach der das Wasserstoffpotential von [H3O+] abhängt, also pH-abhängig ist. Das Prinzip der Methode wird im folgenden Kapitel behandelt. Für einen Versuchsaufbau wie der Normalwasserstoffelektrode gilt wegen E° = 0 und wegen pH = −lg [H3O+]:
E/V = 0,059 · lg [H3O+] = −0,059 · pH
Die gegen eine Normalwasserstoffelektrode gemessene Potentialdifferenz der Wasserstoffelektrode in der Messlösung ist so dem pH-Wert proportional. Die Linearität der Beziehung erlaubt präzise Messungen nach einer unaufwendigen 2-Punkt-Eichung. In der Praxis werden anstelle der Normalwasserstoffelektrode leichter handhabbare „Einstabmessketten“ verwendet.
Zahlreiche Zusammenhänge in diesem umfangreichen Kapitel lassen sich durch eine graphische Darstellung aufzeigen. Im oberen Teil des folgenden Bildes ist eine Speziesverteilungskurve für das Phosphorsäuresystem gezeigt. Beginnend mit reiner Phosphorsäure auf der linken Seite des Diagramms lässt sich die fortschreitende Bildung deprotonierter Spezies beim Erhöhen des pH-Wertes verfolgen.
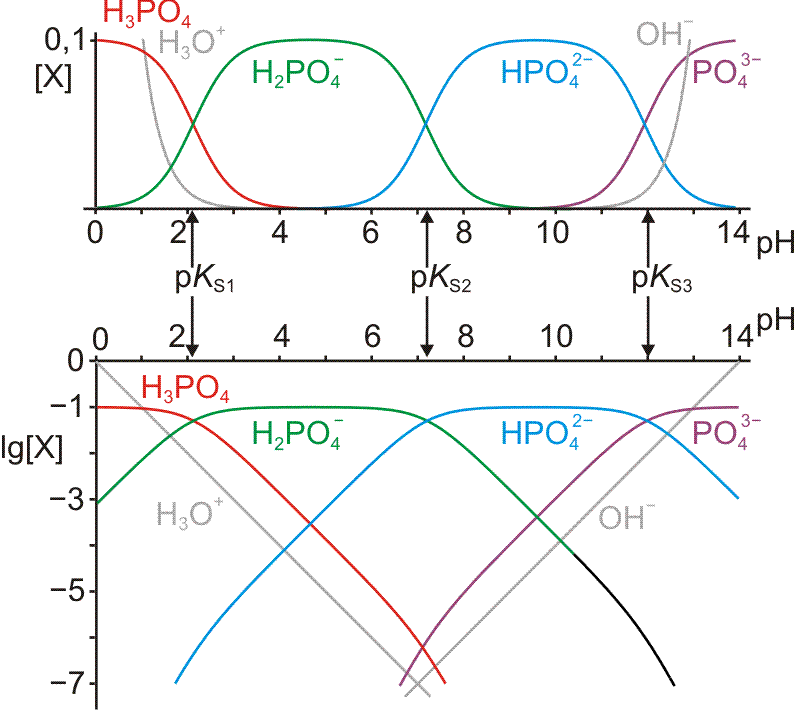
Spezies in einer Phosphorsäure-Lösung. Oben: Einfache Speziesverteilungskurve, [X] ist die Konzentration der jeweils betrachteten Spezies; unten: Speziesverteilungskurve mit logarithmischer Skalierung der Ordinate („Hägg-Diagramm“). Verwendete Zahlenangaben: c0 = 0,1 mol L−1. pKS1 = 2,1, pKS2 = 7,2, pKS3 = 12,0. Bei den zugrunde liegenden Rechnungen wurden keine Näherungsgleichungen angewandt.
Für eine direkte graphische Auswertung eignet sich der untere Teil des Bildes erheblich besser. Als Beispiel wird demonstriert, wie eine rechnerisch aufwendige Aufgabe aus dem „Mortimer“ graphisch im Handumdrehen gelöst werden kann. Neben dieser Aufgabe, die nach der Konzentration aller Phosphorspezies in einer Phosphorsäurelösung fragt, lassen sich alle Äquivalenzpunkte der Titration der dreiprotonigen Säure H3PO4 ablesen.
Die Ionen in einer wässrigen Metallsalzlösung sind hydratisiert, das heißt, sie sind von Wasserdipolen eingehüllt. Nicht nur Wassermoleküle, sondern auch andere Teilchen – meist solche, die Brønsted-Basen sind wie OH−, NH3, Säurerest-Anionen (Cl−, Br−, I−, PO43−, CN−) – können sich um Metallionen herumlagern, sie können als Ligand auftreten und Komplexverbindungen mit einem Metallion als Zentralatom bilden.
Viele Brønsted-Basen können also nicht nur eine Bindung zu H+ aufbauen, wobei die konjugierten Säuren entstehen, sondern auch zu anderen positiv geladenen Ionen; die chemische Bindung erfolgt dabei durch ein freies Elektronenpaar aus der Valenzschale der Base (dative Bindung oder koordinative Bindung). Das Lewis-Säure-Base-Konzept ist ein Versuch, die Gemeinsamkeiten zwischen Komplexbildungsreaktionen und Brønsted-Säure-Base-Reaktionen hervorzuheben. Danach ist eine Lewis-Base ein Molekül oder Ion mit einem freien Elektronenpaar in der Valenzschale, eine Lewis-Säure ist ein Teilchen, das eine Elektronenpaarlücke aufweist und so eine dative Bindung von einer Base annehmen kann. Alle Brønsted-Basen sind auch Lewis-Basen, der wesentliche Unterschied tritt bei der Säuredefinition auf: H+ ist ebenso wie Metallionen eine Lewis-Säure, nach Brønsted aber nur das von einer Säure abgegebene Teilchen.
Komplexverbindungen spielten bereits im Vorhergehenden eine Rolle, nämlich außer bei der Bindung von Wasser als Ligand an Metallionen bei der Veränderung der Löslichkeit durch Komplexbildung (Silberhalogenide, Fixieren photographischer Schichten). In der belebten Natur sind roter Blutfarbstoff, Chlorophyll und Vitamin B12 Komplexverbindungen von Eisen, Magnesium und Cobalt. Eine Einführung in die Chemie der Komplexverbindungen wird am Ende von Teil B gegeben, da dieser Verbindungstyp vor allem bei den Nebengruppenelementen von Bedeutung ist.
Eine gerade in diesem Kapitel verwirrende und unglückliche Regelung: die Formeln von Komplexverbindungen werden in eckige Klammern gesetzt, das hydratisierte Aluminium-Ion wird als [Al(H2O)6]3+ bezeichnet. Elektroneutrale Komplexverbindung wie die im Praktikum auftretenden „inneren Salze“ würden dann im Stil [Ni(dmg)2] bezeichnet, womit hier nicht die Konzentration an Ni(dmg)2 gemeint ist.
In der Schreibweise kann man eine dative Bindung hervorheben, indem man für das bindende Elektronenpaar anstelle eines Striches einen Pfeil von der Base zur Säure hin zeichnet. Hierdurch wird ausgedrückt, dass die beiden Bindungselektronen dem Elektronenpaardonator zugerechnet werden, dass also keine Formalladungen durch die dative Bindung entstehen. Alternativ wird in den Lehrbüchern die dative Bindung wie eine normale kovalente Bindung formuliert. Die beiden geläufigen Schreibweisen sehen im Beispiel des „Amminborans“ so aus:
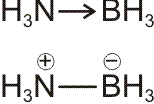
Die Pfeilschreibweise wird umständlich, wenn normale kovalente Bindungen und dative Bindungen gemeinsam in einem Teilchen auftreten. Eine – nicht sehr befriedigende – Möglichkeit ist die Formulierung mesomerer Grenzstrukturen. Ein Beispiel ist das Anion in Kalium-tetrachloridoaluminat:
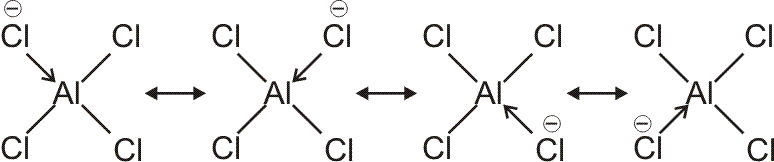
Der Verzicht auf die Pfeilschreibweise führ zu einer merklichen Vereinfachung, allerdings findet sich die negative Formalladung nun auf dem elektropositiven Aluminium wieder:
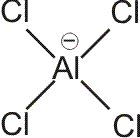
Es stellt sich natürlich die Frage, ob tatsächlich eine der vier Bindungen anders ist als die anderen, oder, allgemeiner, ob die dative Bindung eine normale kovalente Bindung ist, die nur zufällig mit dem Elektronenpaar eines Partners aufgebaut worden ist – in diesem Fall wäre die Mühe mit dem Pfeil sinnlos. Gibt es also einen Unterschied in der zentralen Bindung in den isosteren Molekülen Amminboran und Ethan?
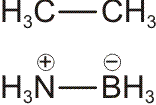
Man beachte, dass die Bildung der Bindung nicht zu einer Unterscheidung führen kann, da Ethan sich aus zwei CH3-Radikalen genauso bildet wie aus Verbindungen, die als Quelle für ein CH3−- und ein CH3+-Ion dienen. Entscheidend ist vielmehr die Art der Bindungsspaltung bei Zufuhr der Bindungsdissoziationsenergie D. Es war gezeigt worden, dass alle „normalen“ chemischen Bindungen – kovalente Bindungen genauso wie Ionenbindungen – durch D homolytisch gespalten werden. Ethan zerfällt daher bei der Spaltung mit der niedrigst möglichen Energie in zwei Methyl-Radikale. Die dative Bindung in Amminboran hingegen spaltet heterolytisch. Dies führt zu einer klaren Unterscheidung zwischen dativer und normaler Bindung. Mit diesem Ergebnis in Einklang sind weitere Unterschiede, so ist die kovalente Bindung in Ethan ca. dreimal so stark wie die dative Bindung in Amminboran.
Und was ist mit [AlCl4]−? Die vier Al-Cl-Bindungen sind natürlich äquivalent (genau das sagen die mesomeren Grenzformeln ja aus!), aber in Übereinstimmung mit der Besonderheit der dativen Bindung wird die erste Bindung heterolytisch, die zweite, dritte und vierte aber homolytisch gespalten, wenn zunehmend mehr Energie zugeführt wird. Diese auf den ersten Blick irritierende Aussage wird klar, wenn man sich bewusst macht, dass [AlCl4]−-Ionen nicht für Experimente zur Verfügung stehen, sondern nur Verbindungen wie Na[AlCl4]. Beim Erhitzen wird eine der vier Bindungen mit recht kleiner Energie heterolytisch gespalten gemäß
Na[AlCl4] → NaCl + AlCl3
und erst dann folgt bei deutlich höherer Energie der homolytische Bruch der kovalenten Bindungen im AlCl3. – Welche Schreibweise ist nun angebracht? Da es keine verbindliche Festlegung der IUPAC gibt und der übliche Gebrauch in Lehrbüchern eher lax ist, werden auch wir die Bindung in einem Lewis-Säure-Base-Addukt mit einem einfachen Strich formulieren, trotzdem aber im Hinterkopf wissen, was eine dative Bindung ist.
Die Lewis-Acidität ist mit der positiven Ladungsdichte der Säure korreliert. Die Fähigkeit zur festen Bindung einer Lewis-Base, zum Beispiel des Dipolmoleküls Wasser, ist hoch, wenn die Ladung eines Lewis-aciden Kations hoch ist und/oder wenn sein Radius klein ist (man vergleiche die Faktoren, welche die Gitterenergie einer Ionenverbindung bestimmen). Bindet eine starke Lewis-Säure Wassermoleküle in einer Hydrathülle, so kommt es zu einer abstoßenden Wechselwirkung mit den positiv polarisierten H-Atomen der Wasser-Moleküle – die Lewis-Säure-gebundenen Wassermoleküle sind stärker Brønsted-acide als freie Wassermoleküle. Anders betrachtet: eine hohe Lewis-Acidität setzt sich in eine hohe Brønsted-Acidität um, wenn die anbindende Lewis-Base protolysierbare H-Atome aufweist. Diese Betrachtung erklärt, warum eher große und nicht hoch geladene Lewis-Säuren wie Na+ die Acidität ihrer Hydrat-Wassermoleküle nur so wenig beeinflussen (pKS-Wert des hydratisierten Na+-Ions: 14,7), dass diese Kationen keinen Beitrag zum pH-Wert einer wässrigen Lösung leisten (wenn Sie unsicher sind: … die auch nicht in umfangreicheren pKS-Wert-Tabellen zu finden sind).
Die Erhöhung der Brønsted-Acidität eines Moleküls bei der Bindung an eine Lewis-Säure kann drastisch sein. Der pKS-Wert von Wasser beträgt 15,7, der pKS-Wert des [Fe(H2O)6]3+-Ions 2,4. Die Säurekonstante des Wassers sinkt also um mehr als 13 Zehnerpotenzen, wenn dieses an ein Fe3+-Ion bindet – denken Sie doch schon einmal über die Acidität des [P(H2O)6]5+-Ions nach.
Während bei Säure-Base-Reaktionen ein Ungleichgewicht in der Elektronenverteilung ausgeglichen wird, ohne dass Elektronen oder Elektronenpaare vollständig von einem Reaktionspartner auf den anderen übertragen werden, geschieht bei einer Redox-Reaktion genau das. Bei einer Reaktion wie der von Natrium mit Chlor zu Natriumchlorid ist der Elektronenübergang offensichtlich. Bei der Reaktion von Wasserstoff mit Chlor zu Chlorwasserstoff und dessen anschließender Protolyse in Wasser entsteht aber in zwei Schritten auch Chlorid – aber wo genau ist hier der Elektronenübergang?
| Mortimer: 14 Binnewies: 11.1, 11.2 Riedel: 3.8 |
Versuch 12-7: ZnS-Synthese
Versuch 5-22: CuO + H2
Die Versuche haben eine Gemeinsamkeit: zwischen den Reaktionspartnern werden Elektronen übertragen:
Zn + S → ZnS
CuO + H2 → Cu + H2O
Auch beim letzten Beispiel ist der Elektronenübertrag offensichtlich: Kupfer-Ionen in der gemäß Cu2+O2− aufgebauten Ionenverbindung haben jeweils zwei Elektronen erhalten, wenn anschließend elementares Kupfer vorliegt. Nach alter Sprechweise ist der Entzug von Sauerstoff eine Reduktion, eine Oxidation ist dagegen die Aufnahme von Sauerstoff (daher der Name). Magnesium wurde bei der Verbrennung des Metalls an der Luft oxidiert:
2 Mg + O2 → 2 Mg2+O2−
Vom Standpunkt des Magnesiums aus verläuft die Umsetzung des Metalls mit Chlor völlig analog:
Mg + Cl2 → Mg2+Cl−2
Oxidation wird daher allgemeiner als Entzug von Elektronen definiert, Reduktion als Aufnahme von Elektronen. Es werden also Elektronen von einem Reaktionspartner auf den anderen übertragen, es findet eine Redox-Reaktion statt. Der Partner, der den anderen oxidiert und dabei selbst reduziert wird, ist das Oxidationsmittel, der Partner, der den anderen reduziert und dabei selbst oxidiert wird, ist das Reduktionsmittel. Man beachte die Verwandtschaft der Konzepte: bei einer Redox-Reaktion werden Elektronen zwischen zwei Partnern ausgetauscht, bei der Protolyse Protonen. In beiden Fällen steht der Austausch im Mittelpunkt, nicht die isolierte Teilreaktion, die nur auf dem Papier formuliert werden kann.
Um die Elektronenbilanz auch bei einer Reaktion wie der Reduktion der Cu2+-Ionen durch Wasserstoff aufstellen zu können, ist ein Konzept nötig, mit dessen Hilfe auch nichtionische Stoffe in die Betrachtung einbezogen werden können. Dies wird durch das Konzept der Oxidationszahl geleistet.
Im einfachen Fall einer Ionenverbindung wie NaCl ist die Ladung der Ionen gleich der Oxidationszahl der Atome. Im Gegensatz zur Ionenladung werden Oxidationszahlen jedoch auch bei kovalenten Bindungen zugeordnet. Man versteht dabei unter der Oxidationszahl eines Atoms dessen Ladung, wenn man sich die betreffende Verbindung aus Ionen aufgebaut denkt. Hierzu ordnet man bindende Elektronenpaare dem elektronegativeren Bindungspartner zu. Aufgrund dieser Definition sind Oxidationszahlen nur bei Ionenverbindungen auch reale Ladungen, bei kovalenten Verbindungen sind die Ladungsangaben fiktiv. Der Sinn dieses Formalismus ist offensichtlich: bei der Formulierung einer Redox-Reaktion liegt nun ein Werkzeug vor, dass die Zuordnung von Ladungen zu einzelnen Atomen erlaubt. Es wird so die Situation vermieden, dass über mehrere Atome delokalisierte Elektronen nur unbequem und fehlerträchtig bilanziert werden können. In der Schreibweise werden Oxidationszahlen von Ionenladungen unterschieden. Eine Ionenladung wird als arabische Zahl mit nachgestelltem Vorzeichen geschrieben: 2−, eine Oxidationszahl durch Vorzeichen und römische Zahl: −II.
Man beachte dabei den Unterschied zwischen der formalen heterolytischen Spaltung von Bindungen bei der Bestimmung der Oxidationszahlen und der homolytischen Spaltung zur Berechnung von Formalladungen. Bindungen zwischen gleichartigen Atomen sind unpolar. Bei der Berechnung der Oxidationszahlen werden in diesem Fall die Bindungselektronen zu gleichen Teilen auf die Bindungspartner aufgeteilt – nur in diesem Fall werden auch hier die Bindungen homolytisch gespalten. Elemente erhalten so stets die Oxidationszahl 0.
Um mit Hilfe der angegebenen Definition die Oxidationszahlen in einer Verbindung aus mehr als zwei Elementen zu bestimmen, muss der Aufbau der Verbindung bekannt sein. Es gelten jedoch meist einige Regeln, die es erlauben, die Oxidationszahlen nach einem einfachen Schema zu bestimmen:
1. Bei einatomigen Teilchen (Atomen, Ionen) ist die Oxidationszahl gleich der Ladung des Teilchens,
2. Bindungselektronen zwischen gleichartigen Atomen werden zu gleichen Teilen zwischen diesen verteilt; Elemente erhalten die Oxidationszahl 0,
3. Die Summe der Oxidationszahlen ist 0 für eine Verbindung und gleich der Ionenladung für ein mehratomiges Ion,
4. einige Elemente kommen meist mit nur einer Oxidationszahl vor:
a) Fluor besitzt als elektronegativstes Element in allen Verbindungen die Oxidationszahl −I,
b) Sauerstoff hat in den meisten Verbindungen die Oxidationszahl −II (Ausnahme: Peroxide),
c) In Verbindungen der Elemente der 1. und 2. Hauptgruppe des Periodensystems (Alkali- und Erdalkali-Elemente) besitzen diese die Oxidationszahlen I bzw. II,
d) Wasserstoff besitzt in den meisten Verbindungen die Oxidationszahl I (Ausnahme: Metallhydride wie NaH mit H−-Ionen).
Mit Hilfe dieser Regeln ergeben sich die Oxidationszahlen der Atome in vielen Verbindungen und Ionen. Stellen Sie fest, ob Sie ein Problem haben bei: NaCl, MgCl2, SO42−, PO43−, Na3PO4, K2Cr2O7, FeO, Fe2O3; NH3, N2H4, NH2OH, N2, N2O, NO, N2O3, NO2, HNO3.
Ein Hauptgruppenelement kann bei der Bildung einer Verbindung höchstens alle seine N Valenzelektronen, also der Gruppennummer −10, für Bindungen zur Verfügung stellen. Für die Oxidationszahlen gelten daher: Die höchste Oxidationszahl eines Hauptgruppenelementes entspricht seiner Valenzelektronenzahl N. Die niedrigste Oxidationszahl erreicht es, wenn es seine Valenzschale bis zur nächsten Edelgaskonfiguration auffüllt: Die niedrigste Oxidationszahl eines Hauptgruppenelementes ist N − 8. Die vorstehende Reihe der Stickstoffverbindungen zeigt dieses Prinzip bei Ammoniak und Salpetersäure; weitere Beispiele sind H2S und H2SO4; HCl and HClO4.
Nach Einführung der Oxidationszahlen kann alternativ definiert werden: Oxidation ist nun auch die Erhöhung der Oxidationszahl, Reduktion deren Erniedrigung. Negativ ausgedrückt: eine Reaktion ohne Oxidationszahländerung ist keine Redox-Reaktion – diese Regel gilt streng und hilft, manchen Irrtum zu vermeiden (wir diskutieren ein paar schlimme Fälle, die man gar nicht aufschreiben mag).
Mit Hilfe der Oxidationsstufen kann die formale Ähnlichkeit von Protolyse und Redox-Reaktion weiter vertieft werden:
Versuch 14.1: Reaktion von KBr und KI mit Chlorwasser, KI mit Bromwasser • Chlor setzt aus Bromid-Lösung Brom und aus Iodid-Lösung Iod frei; das freigesetzte Brom kann durch Ausschütteln in Benzin gewonnen werden; bei der anschließenden Reaktion mit Iodid setzt Brom Iod frei.
Eine Reaktionsgleichung zum Versuch lautet:
Cl±02 + 2 Br− → 2 Cl− + Br±02
Die Reaktion lässt sich als Umsetzung zweier Redoxpaare verstehen: 2 Cl−/Cl2 und 2 Br−/Br2, analog zur gedanklichen Zerlegung eines Protolysegleichgewichts in zwei konjugierte Säure-Base-Paare.
Sind Ausgangsstoffe und Endprodukte einer Redox-Reaktion bekannt(!), so kann eine Reaktionsgleichung, hier eine Redox-Gleichung, aufgestellt werden werden. Hierzu sind die stöchiometrischen Faktoren zu berechnen; zweckmäßigerweise wird für die beiden Redoxpaare getrennt formuliert und anschließend werden die erhaltenen Teilgleichungen unter Berücksichtigung der Elektronenbilanz verknüpft. Da dies ein typischer Gegenstand von Übungen ist, soll nur das Prinzip an einem Beispiel aufgezeigt werden:
Versuch 21-11: Kupfer und Salpetersäure • Kupfermetall löst sich in konzentrierter Salpetersäure unter Bildung von Kupfer(II) und NO2, während mit halbkonzentrierter Säure NO anstatt NO2 entsteht.
Beim Aufstellen der Teilgleichungen für die beiden Redoxpaare wird in der folgenden Weise vorgegangen (als Beispiel dient die Auflösung von Kupfer in halbkonzentrierter Salpetersäure, wobei Kupfer(II)-Ionen und Stickstoffmonoxid entstehen. Da Salpetersäure eine starke Säure ist, wird mit der protolysierten Form H3O+ + NO3− formuliert; dies ist kein kritischer Punkt, wer mag, kann auch von HNO3 ausgehen).
Die Teilgleichung für das Redoxpaar 1 ist problemlos aufzustellen:
Cu → Cu2+ + 2 e−
Die Teilgleichung für das Redoxpaar 2 wird auf folgende Weise entwickelt:
Das redoxaktive Element: die Oxidationszahlen der beteiligten Atome zeigen, bei welchem Element eine Änderung eingetreten ist (hier bei N):
NVO−II3− → NIIO−II
Die Elektronenzahl: die Differenz der Oxidationszahlen ist die Zahl der Elektronen, welche die reduzierte Form mehr besitzt als die oxidierte Form:
NVO3− + 3 e− → NIIO
Die Ladungsbilanz: die Summe der Ladungen auf jeder Seite der Gleichung muss gleich sein. Auf die Seite mit überschüssiger negativer Ladung werden im Fall einer sauren Lösung H3O+-Ionen zugefügt, im Fall einer basischen Lösung werden OH−-Ionen auf die Seite mit geringerer negativer Ladung zugefügt:
NVO3− + 3 e− + 4 H3O+ → NIIO
Die Stoffbilanz wird ausgeglichen. Die Zahl der Atome jeder Atomsorte muss auf beiden Seiten der Gleichung gleich sein; der Ausgleich erfolgt durch Wasser:
NVO3− + 3 e− + 4 H3O+ → NIIO + 6 H2O
Fertig. Nun kommmt noch die Kombination der Teilgleichungen; hierzu werden die beiden Teilgleichungen so mit Faktoren multipliziert, dass die Elektronenzahlen in beiden Teilgleichungen gleich sind (kleinstes gemeinsames Vielfaches der Elektronenzahlen der Teilgleichungen); die Redoxgleichung wird dann durch Addition der beiden Teilgleichungen mit jetzt gleicher Elektronenzahl erhalten:
Cu → Cu2+ + 2 e− × 3
NVO3− + 3 e− + 4 H3O+ → NIIO + 6 H2O × 2
3 Cu + 2 NVO3− + 6 e− + 8 H3O+ → 3 Cu2+ + 6 e− + 2 NIIO + 12 H2O
oder, nach Subtrahieren der auf beiden Seiten auftretenden Teilchen, hier der 6 e−:
3 Cu + 2 NO3− + 8 H3O+ → 3 Cu2+ + 2 NO + 12 H2O
Ist es zum Beispiel für stöchiometrische Berechnungen wichtig, die Ionengleichung in eine Substanzgleichung umzuwandeln – es könnte zum Beispiel gefragt sein, wieviel Gramm Kupfer(II)-nitrat man aus 1 kg Kupfer herstellen kann –, so sollte dies erst jetzt erfolgen, nachdem der Redox-Vorgang vollständig beschrieben ist, zum Beispiel (8 HNO3 statt 8 H3O+, dafür links 2 NO3− und rechts 8 H2O einsparen):
3 Cu + 8 HNO3 → 3 Cu(NO3)2 + 2 NO + 4 H2O
… oder, wenn festes Kupfer(II)-nitrat-Trihydrat gewonnen werden soll:
3 Cu + 8 HNO3 + 5 H2O → 3 Cu(NO3)2·3 H2O + 2 NO
Beachten Sie die Schreibweise bei Additionsverbindungen: die erste „3“ bei 3 Cu(NO3)2·3 H2O wirkt auch auf die Kristallwassermoleküle, es geht also um 9 H2O!
Energie bedeutete bisher fast immer Wärmeenergie – bei einer endothermen Reaktion war sie ständig zuzuführen, bei einer exothermen wurde sie frei (genau das meint „…therm“). Eine endotherme Reaktion durch elektrische Energie zu erzwingen eröffnet viele neue Möglichkeiten, ebenso wie es eine interessante Aufgabe ist, eine exotherme Reakion so auszuführen, dass elektrische Energie gewonnen werden kann.
| Mortimer: 21 Binnewies: 11.3-11.10 Riedel: 3.8 |
Versuch 12-4: Elektrolyse von LiBr-Schmelze • LiBr leitet nur im geschmolzenen Zustand den elektrischen Strom. Bei der Elektrolyse entsteht gemäß 2 LiBr → 2 Li + Br2 neben Lithiummetall auch elementares Brom, Br2, das bei der hohen Temperatur der Schmelze aus dieser herausdampft. Dieses entweichende Brom kann mit einem Iodid-getränkten Filterpapier nachgewiesen werden, da Brom Iodid zu Iod oxidieren kann. Um den Nachweis empfindlich zu gestalten, ist neben Iodid Stärke vorhanden, die mit dem entstehenden Iod einen blauen Einschlusskomplex (siehe später im Kapitel „Halogene“) bildet.
Versuch 21-12b: Elektrolyse von Zinkiodid in wässriger Lösung • die Elektrolyse führt gemäß ZnI2 → Zn + I2 zur Bildung von elementarem Iod, das durch seine dunkelbraune Farbe auffällt. Nach dem Abschalten der Elektrolyse wirkt die Elektrolysezelle als Batterie; es bleibt für den Augenblick offen, wieso.
Werden Elektroden in eine Elektrolytlösung, im Versuch Zinkiodid in Wasser, eingetaucht, so folgen die Ionen dem elektrischen Feld. Ist die angelegte Spannung hinreichend groß, so werden die Ionen beim Erreichen der Elektrode entladen (an der Anode oxidiert, an der Kathode reduziert). Im Beispiel werden Zn2+-Ionen an der der Kathode gemäß
Zn2+ + 2 e− → Zn
zu Zinkmetall reduziert. Die Abscheidung eines Stoffes mit Hilfe des elektrischen Stroms heißt Elektrolyse. Für die bei einer Elektrolyse abgeschiedene Stoffmenge gelten die Faradayschen Gesetze:
• Die Masse eines elektrolytisch abgeschiedenen Stoffes ist der durch den Elektrolyten geflossenen Ladungsmenge direkt proportional.
• Die durch gleiche Ladungsmengen abgeschiedenen Massen verschiedener Stoffe verhalten sich wie die Äquivalentmassen dieser Stoffe zueinander (Äquivalentmasse = Atommasse/z mit z = Zahl der aufgenommenen bzw. abgegebenen Elektronen).
Um 1 Grammäquivalent (= 1 mol/z) eines Stoffes abzuscheiden, wird eine Ladungsmenge von ca. 96500 C benötigt:
NA · e = 6,022 · 1023 · 1,6021 · 10−19 C = 96524 C
Diese Ladungsmenge, die 1 mol Elektronen entspricht, wird 1 F (Faraday) genannt. 1 F scheidet zum Beispiel aus den entsprechenden Metallsalzlösungen oder -schmelzen ab:
aus AgI-Salzlösung 107,87/1 g = 107,87 g Silber,
aus CuII-Salzlösung 63,54/2 g = 31,77 g Kupfer,
aus AlIII-Salzschmelze 26,98/3 g = 8,99 g Aluminium.
Während bei der Elektrolyse eine freiwillig nicht ablaufende Reaktion durch Energiezufuhr erzwungen wird, gewinnt man bei einem galvanischen Element (Batterie, Akkumulator) die bei einer Redoxreaktion freiwerdende Energie in Form von elektrischer Energie. Eine geeignete Redoxreaktion ist die Reduktion von Cu2+-Ionen (zum Beispiel aus Kupfersulfat) durch Zink zu Kupfer, wobei Zn2+-Ionen entstehen:
Cu2+ + Zn → Cu + Zn2+
Versuch 21-1: Reaktion von Zink mit Kupfer(II)-Lösung
Das Daniellelement ist eine Anordnung, bei der die Redoxpaare Zn/Zn2+ und Cu/Cu2+ so gekoppelt sind, dass die bei der Reaktion freiwerdende Energie nicht als Wärme anfällt, sondern als elektrische Energie.
Versuch 21-8: Daniellelement
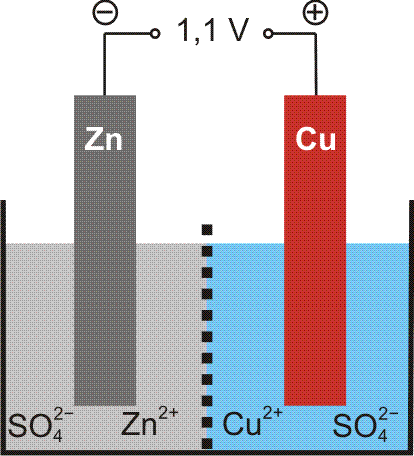
Aus der Lage des Redoxgleichgewichts folgt, dass Zink ein größeres Bestreben als Kupfer hat, Elektronen abzugeben, es ist ein stärkeres Reduktionsmittel. Dementsprechend ist Zink im Daniellelement der Minus-Pol, also die Elektrode mit dem höheren Elektronendruck. Besteht eine leitende Verbindung zum Kupferstab, so fließen Elektronen von Zink zu Kupfer. Der Kupferstab steht in Kontakt mit Cu2+-Ionen in der Kupfersulfat-Lösung, die durch die herüberfließenden Elektronen reduziert werden. Auf dem Kupferstab scheidet sich weiteres Kupfer ab. Da am Zinkstab eine entsprechende Zahl Zn2+-Ionen in Lösung geht, entsteht – bezogen auf die Zahl der Anionen – ein Überschuss von Kationen auf der Zinkseite und ein Unterschuss auf der Kupferseite. Der Ladungsausgleich zwischen den Lösungen erfolgt durch eine poröse Wand zwischen den beiden Halbzellen, die eine Durchmischung der Lösungen behindert, aber die Wanderung der Anionen im elektrischen Feld zulässt. Die in den Halbzellen ablaufenden Reaktionen sind die Teilreaktionen der Redoxreaktion:
Oxidation: Zn → Zn2+ + 2 e−
Reduktion: Cu2+ + 2 e− → Cu
Gesamtreaktion: Cu2+ + Zn → Cu + Zn2+
Die ohne Stromfluss gemessene Spannung des galvanischen Elementes ist die Elektromotorische Kraft (EMK) der Zelle. Aufgrund dieser Potentialdifferenz kann die galvanische Zelle Arbeit leisten.
Versuch 21-6. Potentiale von Zink, Zinn, Kupfer, Silber
Die EMK einer Zelle ist die Differenz der Potentiale der beiden Halbzellen, ΔE; wird unter Standardbedingungen gemessen, ergibt sich ΔE°. Einzelpotentiale von Halbzellen lassen sich ebensowenig messen, wie die zugrundeliegende Teilreaktion der Redoxreaktion nicht ohne Kopplung an ein zweites Redoxpaar ablaufen kann. Um Potentialdifferenzen zwischen beliebigen Kombinationen zweier Halbzellen berechnen zu können, wurde daher eine Standard-Halbzelle definiert und deren Potential zu 0 V festgelegt (vergleiche die Definition von Standardbildungsenthalpien mit willkürlicher Festlegung des Wertes 0 kJ mol−1 für Elemente in der stabilsten Form). Als Standard-Halbzelle wurde die Normalwasserstoffelektrode (NWE) eingeführt. Hierbei handelt es sich um eine Halbzelle, bei der eine platinierte Platinelektrode (mit feinverteiltem Platin überzogene Platinelektrode) in eine Lösung von H3O+-Ionen taucht und mit H2-Gas umspült wird; gemessen wird unter Standardbedingungen: [H3O+] = 1, p(H2) = 1 atm = 1,013 bar, T = 25 °C = 298 K. Das Normalpotential (Standardpotential) E° einer Halbzelle ist die EMK der galvanischen Zelle, die sich bei der Kombination dieser Halbzelle mit der NWE ergibt. Auch für die zu messende Halbzelle werden Standardbedingungen eingehalten: [gelöste Reaktanden] = 1, p(Gase) = 1 atm, T = 25 °C. Normalpotentiale sind also Relativwerte bezogen auf die NWE mit einem auf 0 V festgelegtem Potential.
Gibt ein Halbelement Elektronen an die NWE ab, findet also eine Reduktion der NWE statt, so erhält das gemessene Potential ein negatives Vorzeichen. Wird die NWE hingegen durch ein Halbelement oxidiert, wenn also die NWE als Reduktionsmittel wirkt, so erhält das Potential ein positives Vorzeichen. Werden die Normalpotentiale der verschiedenen Redoxpaare nun nach steigendem Zahlenwert geordnet, so erhält man die elektrochemische Spannungsreihe (siehe Anhang). Die Normalpotentiale erlauben eine Vorhersage, ob eine Redox-Reaktion in wässriger Lösung stattfinden wird. Je höher ein Redoxpaar in der Spannungsreihe steht, ein umso stärkeres Reduktionsmittel ist die reduzierte Form; entsprechend stellen die oxidierten Formen der weiter unten stehenden Redoxpaare zunehmend stärkere Oxidationsmittel dar. Ein Redoxvorgang wird dann stattfinden, wenn eine Kombination „rechts plus darunter links“ gewählt wird. Ein Beispiel ist die Reaktion des Daniell-Elements
| ox. | red. | n e− | E°/V |
|---|---|---|---|
| Zn2+ | Zn | 2 | −0,76 |
| Cu2+ | Cu | 2 | +0,35 |
Dieses einfache Verfahren führt bei hinreichend großen Potentialdifferenzen zu einer richtigen Aussage, da dann eine Abweichung der Konzentrationen von den Standardbedingungen eher vernachlässigt werden darf. Die Potentialdifferenz zweier Redoxpaare (im Beispiel: +0,35 − (−0,76) = 1,11 V) ist die EMK der entsprechenden galvanischen Zelle.
Versuch 21-1: Reaktion verschiedener Metalle mit verschiedenen Metallsalzlösungen
Auf Reaktionen mit starken Säuren im Konzentrationsbereich von ca. 1 mol L−1 angewandt, ergibt sich, dass die über Wasserstoff stehenden Elemente (E° < 0) unter Bildung von H2 aufgelöst werden, zum Beispiel:
Zn + 2 H3O+ → Zn2+ + H2 + 2 H2O
Die unter Wasserstoff stehenden Metalle (E° > 0) hingegen lösen sich in Säuren nicht unter Wasserstoffentwicklung. So reagiert Kupfer nicht mit Salzsäure; beim Auflösen von Kupfer in Salpetersäure entsteht nicht Wasserstoff, sondern Stickstoffmonoxid (siehe voriges Kapitel):
Maßgeblich ist hier das Normalpotential des Redoxpaares NO/NO3−; die Oxidation von Kupfer durch H3O+ ist dagegen nach der Spannungsreihe nicht zu erwarten:
| ox. | red. | n e− | E°/V |
|---|---|---|---|
| H3O+ | H2 + 2 H2O | 2 | 0 |
| Cu2+ | Cu | 2 | +0,35 |
| NO3− + 4 H3O+ | NO + 6 H2O | 3 | +0,96 |
Die Auflösung von Kupfer in Salpetersäure steht also im Einklang mit der genannten Regel. Die in der Spannungsreihe unter Wasserstoff stehenden Metalle (E° > 0) werden wegen ihrer Beständigkeit gegen nichtoxidierende Säuren (= Säuren, in denen kein stärkeres Oxidationsmittel als H3O+ vorliegt) Edelmetalle genannt, die darüberstehenden Metalle werden als „unedel“ bezeichnet.
Eine chemische Reaktion läuft freiwillig ab, wenn die Änderung der freien Enthalpie negativ ist (ΔG < 0). Da ΔG die maximale Nutzarbeit einer Reaktion ist, kann diese Größe mit ΔE einer galvanischen Zelle verknüpft werden. Die elektrische Arbeit der Zelle ist das Produkt aus geflossener Ladung und Spannung, die maximale Spannung der Zelle ist die EMK. Für das Daniell-Element mit einer EMK von 1,10 V ergibt sich so die maximale elektrische Arbeit A für 1 mol Formelumsatz zu:
A = 2 F ΔE = 2 · 96485 C mol−1 · 1,10 V = 212 · 103 CV mol−1 = 212 kJ mol−1
da wegen Cu2+ + Zn → Cu + Zn2+ die Ladung 2 F fließt. Die errechnete Arbeit ist die maximale Nutzarbeit, die vom System geleistet werden kann (ΔG = −212 kJ mol−1), es ist also mit der geltenden Vorzeichenkonvention:
A = ΔG = − n F ΔE
Im Beispiel wurde der Zahlenwert für E° verwendet, es gilt also:
ΔG° = − n F ΔE°
Um das Vorzeichen von ΔE° richtig zu bestimmen, wird die folgende Potentialdifferenz gebildet:
ΔE° = E°Reduktion − E°Oxidation
Bei der Redoxreaktion des Daniellelements wird Cu2+ reduziert und Zn oxidiert. Es gilt daher:
ΔE° = E°(Cu/Cu2+) − E°(Zn/Zn2+) = 0,34 V − (−0,76 V) = 1,10 V
ΔG° zeigt durch seinen negativen Zahlenwert, dass die Reaktion in dieser Richtung freiwillig abläuft, während die umgekehrte Reaktion – die Elektrolyse − mit ihrem positiven ΔG-Wert durch Energiezufuhr erzwungen werden muss. Wegen
ΔG = −RT ln K
können weiterhin Gleichgewichtskonstanten durch elektrochemische Messungen bestimmt werden. Es gilt mit Zehnerlogarithmen:
ΔG° = − n F ΔE° = −2,303 RT lg K
| ΔE° = | 2,303 RT | lg K |
| nF |
Für T = 25 °C = 298,15 K gilt dann:
| ΔE°/V = | 0,059 | lg K |
| n |
Die Vorhersage einer Redoxreaktion mit Hilfe der Spannungsreihe führt nicht immer zum richtigen Ergebnis. Wird Aluminium oder Eisen mit konzentrierter Salpetersäure versetzt, so wäre die Auflösung des Metalls zu erwarten:
| ox. | red. | n e− | E°/V |
|---|---|---|---|
| Al3+ | Al | 3 | −1,69 |
| Fe2+ | Fe | 2 | −0,41 |
| NO3− + 4 H3O+ | NO + 6 H2O | 3 | +0,96 |
Die Reaktion sollte aufgrund der großen Potentialdifferenzen von 2,65 V und 1,37 V heftig verlaufen.
Versuch 21-14: Passivierung von Eisen durch konzentrierte Salpetersäure, Reaktion mit Kupfersulfat.
Versuch-neu: Reaktion von Eisen mit konzentrierter und verdünnter Salpetersäure
Trotzdem löst sich das Metall nicht auf – es ist passiviert worden. Es hat sich eine Oxidschicht (hier: Aluminiumoxid, Al2O3, bzw. Eisenoxid) gebildet, die durch Einwirkung des starken Oxidationsmittels Salpetersäure gebildet wird und den Rest des Metalls vor dem weiteren Angriff der Säure schützt (vergleiche die „Eloxierung“ von Aluminium). Die Passivierung stellt sich nicht in verdünnter Säure ein (in Stahltanks kann nur konzentrierte Salpetersäure transportiert werden, nicht aber verdünnte Säure).
Versuch 21-10: Potentialdifferenz verschieden konzentrierter Kupfersulfat-Lösungen
Wenn die Konzentrationen der beteiligten Reaktanden von den Standardbedingungen abweichen, wird das Potential eines Redox-Paares mit Hilfe der Nernstschen Gleichung errechnet. Diese ergibt sich aus der freie-Enthalpie-Beziehung, aus der bereits das Massenwirkungsgesetz abgeleitet wurde:
| ΔG = ΔG° + R T ln | [X]x [Y]y |
| [A]a [B]b |
mit ΔG = − n F ΔE und ΔG° = − n F ΔE° zu:
| ΔE = ΔE° − | RT | ln | [X]x [Y]y |
| nF | [A]a [B]b |
Bei Standardtemperatur ergibt sich mit Zehnerlogarithmen und dem Betrag der Potentialdifferenzen in Volt:
| ΔE/V = ΔE°/V − | 0,059 | lg | [X]x [Y]y |
| n | [A]a [B]b |
Angewendet auf einen Teilprozess einer Redoxreaktion, also auf eine Halbzelle, wird erhalten:
| ΔE/V = ΔE°/V − | 0,059 | lg | [red] |
| n | [ox] |
oder
| ΔE/V = ΔE°/V + | 0,059 | lg | [ox] |
| n | [red] |
Bei Standardbedingungen gilt: [ox] = [red] = 1, so dass E = E°. Die Nernstsche Gleichung beschreibt durch den additiven Term 0,059/n · lg([ox]/[red]) die Konzentrationsabhängigkeit von Potentialen. Dadurch ist es möglich, die Vorhersage von Redoxvorgängen auch bei Bedingungen vorzunehmen, die stärker von den Standardbedingungen abweichen.
Bei Metallen und Gasen wie Wasserstoff ist es nicht möglich, in der Lösung als Standardkonzentration 1 mol L−1 einzustellen. Die Konzentration der reduzierten Form in der Lösung ist sehr viel kleiner und meist nicht messbar. In diesem Fall wird jedoch davon ausgegangen, dass sich bei allen Versuchen in wässriger Lösung diese Konzentration nicht ändert, dass also für [red] derselbe Wert vorliegt wie bei der Messung des Standardpotentials. Es ist daher [red] = 1 (wie auch schon bisher lese man nicht: 1 mol L−1, sondern 1 mal so groß, wie bei der Messung des Normalpotentials) eingesetzt; die Nernstsche Gleichung vereinfacht sich dann zu
| ΔE/V = ΔE°/V + | 0,059 | lg [ox] |
| n |
Ein Beispiel ist das Potential des Wasserstoffs in Wasser; die Konzentration von H3O+-Ionen in Wasser beträgt 10−7 mol L−1, für E ergibt sich dann:
E/V = E°/V + (0,059/1) · lg 10−7 = 0 − 0,059 · 7 = −0,41
Mit dem deutlich erniedrigten Potential von −0,41 V sind Wasserstoffionen demnach in Wasser ein schlechteres Oxidationsmittel als in saurer Lösung.
Metalle werden durch Bildung eines stabilen Komplexes in wässriger Lösung „unedler“, das heißt, das Potential des Redoxpaares wird kleiner als das Standardpotential. Die Nernstsche Gleichung beschreibt dies quantitativ. Hierzu wird für [ox] die Konzentration der Metallionen in der Form, wie sie unter Standardbedingungen vorliegen eingesetzt, dass heißt, als hydratisiertes Ion – also als Komplexverbindung mit Wasser als Ligand. Bilden nun andere Liganden stabilere Komplexe, in denen die Metallionen – die oxidierte Form des Redox-Paares – fester gebunden sind, so ist [ox], die Konzentration des Aquakomplexes, kleiner. Bei Standardbedingungen gilt [ox]= 1, bei Komplexbildung gilt [ox] < 1; damit ist lg [ox] < 0 und schließlich E < E°.
Ein galvanisches Element kann Arbeit leisten, da in ihm ein Redox-Prozess freiwillig abläuft. Die entsprechende Umkehrreaktion kann durch Zuführen elektrischer Energie erzwungen werden; es findet eine Elektrolyse statt:
Freiwillig (ΔG < 0, galvanisches Element): Cu2+ + Zn → Cu + Zn2+
Erzwungen (ΔG > 0, Elektrolyse): Cu + Zn2+ → Cu2+ + Zn
Im Daniell-Element läuft die Reaktion von links nach rechts ab; die Standard-EMK ist 1,1 V, Zink ist der Minuspol, Kupfer der Pluspol. Wird nun Zink mit dem Minuspol einer Spannungsquelle verbunden und Kupfer mit dem Pluspol, so kann eine Spannung gegen die EMK des Daniell-Elements geschaltet werden. Überschreitet die von außen angelegte Spannung den Betrag von 1,1 V, so kehrt sich der Elektronenfluss, das heißt, die Richtung des Redox-Prozesses, um; es kommt zur Elektrolyse unter Abscheidung von Zink und der Auflösung von Kupfer.
Das Beispiel des umgepolten Daniell-Elements zeigt, dass für eine Elektrolyse eine Mindestspannung, die Zersetzungsspannung, notwendig ist, die gerade der EMK des entsprechenden galvanischen Elements, das heißt, der Differenz der Redoxpotentiale, entspricht. Die Elektrolyse führt im Beispiel zur Auflösung der Kupfer-Anode. Die Auflösung der Anode tritt nicht auf, wenn die Lösung Stoffe enthält, die zusammen mit ihrer oxidierten Form ein Redoxpaar mit kleinerem Normalpotential ergeben als dies für das Anodenmaterial und dessen oxidierter Form gegeben ist. Meist sind dies Anionen, die im elektrischen Feld zur Anode wandern und dort entladen werden. So werden bei der Chloralkali-Elektrolyse (siehe unten) an der Anode Cl−-Ionen zu Cl2 oxidiert und nicht das Elektrodenmaterial Graphit.
Als praktisch unangreifbares Elektrodenmaterial wird Platin und in der Technik vor allem Graphit verwendet. Bei der Verwendung solcher Hilfselektroden bildet sich das galvanische Gegenpotential erst zu Beginn der Elektrolyse. Das folgende Beispiel zeigt dieses Prinzip: In eine wässrige HCl-Lösung, einer Lösung der Ionen H3O+ und Cl− tauchen eine Platin-Elektrode, die als Kathode geschaltet ist, und eine als Anode geschaltete Graphit-Elektrode. Wird nun eine kleine Spannung angelegt, die unter der für Normalbedingungen errechneten EMK des entsprechenden galvanischen Elements liegt, so kommt es zur Bildung von H2 und Cl2, die an der Elektrodenoberfläche adsorbiert bleiben, indem die folgenden Elektrodenreaktionen ablaufen:
Kathode: 2 H3O+ + 2 e− → H2 + 2 H2O
Anode: 2 Cl− → Cl2 + 2 e−
Der gesamte Redox-Vorgang, der durch die Elektrolyse erzwungen werden soll, folgt demnach der Gesamtgleichung:
2 H3O+ + 2 Cl− → Cl2 + H2 + 2 H2O
Die gebildeten Gase Wasserstoff und Chlor bilden mit den am Redox-Vorgang nicht beteiligten Elektroden aus Platin und Graphit eine Wasserstoff- und eine Chlor-Elektrode. Da zu Beginn die Gasmengen (die Partialdrücke) noch klein sind, sind die Potentiale der beiden Halbzellen der Nernstschen Gleichung, die in Analogie zum Massenwirkungsgesetz auch mit Partialdrücken aufgestellt werden kann, entsprechend ebenfalls klein; für die sich an Kathode und Anode aufbauenden Gegenpotentiale gilt:
| EH/V = | 0,059 | lg | [H3O+]2 |
| 2 | p(H2) |
| ECl/V = E°/V + | 0,059 | lg | p(Cl2) |
| 2 | [Cl−]2 |
Die EMK der galvanischen Zelle, ECl − EH, kompensiert gerade die angelegte Spannung. Mit dem Ansteigen der äußeren Spannung wird mehr Wasserstoff und Chlor abgeschieden, die Gasdrücke werden größer, die EMK der galvanischen Zelle wird größer und kompensiert so die größere angelegte Spannung. Bei weiterer Erhöhung der angelegten Spannung wird der Gasdruck so weit steigen, dass der Atmosphärendruck von 1 atm = 1,013 bar erreicht wird; bei T = 298,15 K und Lösungen der Konzentration 1 mol L−1 wird die EMK der Zelle gleich der Standard-EMK, die sich aus der Differenz der Normalpotentiale der Redoxpaare 2 Cl−/Cl2 (E° = +1,36 V) und H2 + 2 H2O/2 H3O+ (E° = 0 V) errechnet (1,36 V). Bei weiterer Erhöhung der angelegten Spannung über 1,36 V, in diesem Beispiel der Zersetzungsspannung, hinaus, entweichen die Gase aus dem Reaktionsraum, es kommt sichtbar zur Elektrolyse.
Die gemessene Zersetzungsspannung übersteigt oft den berechneten Wert. Eine kinetische Hemmung der Elektrodenreaktion – diese ist vor allem dann häufig, wenn Gase beteiligt sind – wird Überspannung genannt; es gilt:
Zersetzungsspannung = Differenz der Redoxpotentiale + Überspannung
Der Betrag der Überspannung hängt ab von (a) dem Elektrodenmaterial, (b) der Oberflächenbeschaffenheit der Elektrode und (c) der Stromdichte an der Elektrode. Eine besonders hohe Überspannung wird für Wasserstoff an Zink-, Blei- und Quecksilber-Elektroden gefunden. So beträgt bei einer Stromdichte von 0,01 A/cm2 die Überspannung von Wasserstoff an einer Quecksilber-Elektrode 1,12 V; an einer platinierten Platin-Elektrode ist die Wasserstoff-Überspannung 0 V. Die Überspannung von Sauerstoff ist an Platin- und Graphit-Elektroden besonders groß. Ein Beispiel ist die Elektrolyse einer wässrigen HCl-Lösung mit einer Graphit-Anode. Diese oxidiert aufgrund der Sauerstoff-Überspannung Chlorid-Ionen zu Chlor (E° = +1,36 V) und nicht Wasser zu Sauerstoff (E° = +1,23 V), wie es aufgrund der Lage der Normalpotentiale zu erwarten wäre (die Anode oxidiert zuerst die Teilchensorte, deren reduzierte Form zu dem Redoxpaar mit dem kleinsten Potential gehört [in der Spannungsreihe oben steht]; analog wird von der Kathode die Teilchensorte reduziert, deren oxidierte Form zu dem Redoxpaar mit dem größtem Potential gehört [bei Metallen: das edelste Metall]).
Versuch 3-6: Bleiakku
Akkumulatoren sind wiederaufladbare galvanische Elemente, bei denen sich die bei der Stromentnahme (Entladen) ablaufenden Reaktionen durch Zufuhr elektrischer Energie (Laden) wieder umkehren lassen. Akkumulatoren besitzen einen Wirkungsgrad von ca. 80–90 %. Der Bleiakkumulator besteht aus einer Blei- und einer Blei(IV)-oxid-Elektrode, die in Schwefelsäure als Elektrolyt tauchen. Der zugrunde liegende Redox-Vorgang ist:
Pb±0 + PbIVO2 + 2 H2SO4 ⇄ 2 PbIISO4 + 2 H2O
Beim Entladen des Akkumulators verläuft die Reaktion von links nach rechts, beim Aufladen in umgekehrter Richtung. Die negative Elektrode bildet sich durch die Oxidation von Blei:
Pb±0 + SO42− → PbIISO4 + 2 e−
Die positive Elektrode entsteht durch die Reduktion von Blei(IV)-oxid:
PbIVO2 + SO42− + 4 H3O+ + 2 e− → PbIISO4 + 6 H2O
Die Spannung des Bleiakkumulators beträgt ca. 2 V. Dies ist mehr als es aufgrund der Differenz der entsprechenden Normalpotentiale zu erwarten ist. Die größere Spannung ergibt sich aus der Nernstschen Gleichung, wenn die gegenüber den Standardkonzentrationen veränderten Werte berücksichtigt werden: [Pb2+] ist durch Bildung des schwerlöslichen PbSO4 stark verringert, [H3O+] ist in der starken, ca. 30-%-igen Säure erhöht; man beachte hierbei, dass die H3O+-Ionen-Konzentration in das Potential der PbO2-Reduktion eingeht und zwar wegen des Koeffizienten 4 in der Teilgleichung mit der 4. Potenz. Die Gleichung für den ablaufenden Redox-Prozess zeigt, dass mit zunehmender Entladung des Akkumulators die Schwefelsäure-Konzentration abnimmt. Der Ladezustand lässt sich daher durch Messen der Säurekonzentration (in der Praxis: der Säuredichte) überprüfen.
Der Ladevorgang ist eine Elektrolyse. Der Lage der Normalpotentiale nach sollte an der Kathode nicht Blei, sondern der edlere Wasserstoff abgeschieden werden (E°(Pb2+/Pb) = −0,13 V), was durch die hohe Überspannung von Wasserstoff am Elektrodenmaterial Blei nicht geschieht. Die notwendige Überspannung setzt saubere Elektroden voraus; nach einer Verunreinigung des Elektrolyten ist ein Bleiakkumulator nicht wieder aufladbar. Bleiakkumulatoren werden als Autobatterien verwendet, in denen meist sechs Zellen in Reihe geschaltet sind, um so eine Spannung von 6 · 2 V = 12 V zu liefern.
Nickel-Cadmium-Akkumulatoren („NC-Akkus“) liefern eine Spannung von ca. 1,3 V. Zugrunde liegt der Redox-Prozess
Cd + 2 NiO(OH) + 2 H2O → Cd(OH)2 + 2 Ni(OH)2
Bei der Stromentnahme laufen an den Elektroden die folgenden Teilreaktionen ab:
Negative Elektrode:
Cd±0 + 2 OH− → CdII(OH)2 + 2 e−
Positive Elektrode:
2 NiIIIO(OH) +2 H2O + 2e− → 2 NiII(OH)2 + 2 OH−
Versuch neu: Leclanché-Element
Die Taschenlampenbatterie (Trockenelement, Leclanché-Element) ist ein nicht wiederaufladbares galvanisches Element. Der negative Pol besteht aus Zink, der positive Pol aus Braunstein (Mangan(IV)-oxid, MnO2) und der Elektrolyt aus einer angedickten Ammoniumchlorid-Lösung (NH4Cl-Lösung). Die Elektrodenreaktionen sind:
Negative Elektrode:
Zn±0 → Zn2+ + 2 e−
Positive Elektrode:
2 MnIVO2 +2 NH4+ + 2e− → 2 MnIIIO(OH) + 2 NH3
Die bei der Stromentnahme ablaufende Redox-Reaktion ist dann:
2 MnIVO2 +2 NH4+ + Zn±0 → Zn2+ + 2 MnIIIO(OH) + 2 NH3
Der Lithiumionenakku zeichnet sich durch eine besonders hohe Spannung von ca. 3,5 V aus. Er enthält aprotische Lösungsmittel wie Propylencarbonat und ein Leitsalz wie LiPF6.
Versuch neu: Lithiumionenakku
Der negative Pol stellt im geladenen Zustand eine Einlagerungsverbindung von Lithium in Graphit dar, der positive Pol ein Metalloxid wie zum Beispiel Cobalt- oder Manganoxid. Der Redoxprozess ist bei der Manganvariante vereinfacht (vereinfacht, da kein Li-freies MnO2 entsteht):
MnO2 + LiCn → LiMnO2 + Cn
Ordnen Sie Oxidationsstufen zu!
Versuch neu: Brennstoffzelle
Bei den bisher behandelten elektrochemischen Spannungsquellen ist stets der gesamte Vorrat an Reaktanden anwesend. Dies ist bei der Brennstoffzelle anders. Dieses galvanische Element stellt lediglich den Reaktionsraum bereit, während die Ausgangsstoffe kontinuierlich zugeführt werden. Im Experiment wurde eine Wasserstoff/Sauerstoff-Zelle gezeigt, bei der die hohe Bildungsenthalpie des Wassers als elektrische Energie frei wird. Aufgrund der alkalischen Bedingungen im Versuch finden die folgenden Elektrodenreaktionen statt:
Negative Elektrode:
H±02 + 2 OH− → 2 H2O + 2 e−
Positive Elektrode:
O±02 + 2 H2O + 4 e− → 4 OH−
Bei der Korrosion von Metallen spielt die Bildung kleiner galvanischer Elemente an der Oberfläche von Metallen eine wichtige Rolle. Korrosion – die Zerstörung von Metallen durch chemische Einflüsse – kann nicht nur durch aggresive Stoffe wie wässrige Säuren oder Gase hervorgerufen werden. Ein elektrochemischer Vorgang, der bei der Korrosion von Bedeutung ist, ist die Bildung von Lokalelementen. Ein Lokalelement entsteht, wenn die Oberfläche eines unedlen Metalls mit einem edleren Metall in Berührung kommt und ein Elektrolyt die Berührungsstelle umgibt. Ein Beispiel ist Zink, das sich trotz seines negativen Normalpotentials nicht in schwach saurer Lösung gemäß
Zn + 2 H3O+ → Zn2+ + H2 + 2 H2O
löst, da das Metall eine hohe Überspannung für Wasserstoff besitzt. Die Überspannung für Wasserstoff ist an Metallen wie Platin oder Kupfer sehr viel kleiner. Berührt man nun das in Säure eintauchende Zinkstück mit einem Platindraht, so setzt sofort rege Wasserstoffentwicklung am Platindraht(!) ein, da die von Zink abgebenen Elektronen in den Pt-Draht fließen und dort ohne kinetische Hemmung auf H3O+-Ionen übertragen werden. Ein Lokalelement kann auch erzeugt werden, indem ein edleres Metall auf dem unedleren durch eine Redox-Reaktion der Art Zn + Cu2+ → Zn2+ + Cu niedergeschlagen wird.
Versuch 21-13: Pt/Zn- und Cu/Zn-Lokalelement
Zur Korrosion führende Lokalelemente entstehen zum Beispiel dadurch, dass ein edleres Metall als Korrosionsschutz auf ein unedleres aufgezogen wird (Zinn auf Eisen), dann aber die Schutzschicht verletzt wird, so dass das unedlere Metall freigelegt wird. Als Elektrolyt genügt Wasser, da dies meist Salze oder Kohlensäure gelöst enthält, so dass eine hinreichend hohe Leitfähigkeit besteht, um das galvanische Element kurzzuschließen; das unedlere Metall löst sich auf, meist unter Entwicklung von Wasserstoff. Durch die Reduktion von H3O+-Ionen bleiben in der Lösung OH−-Ionen zurück, die mit den entstehenden Metall-Ionen häufig schwerlösliche Hydroxide oder wasserhaltige Oxide bilden. Diese Verbindungen bilden jedoch meist einen nur lockeren Überzug über das Metall, so dass kein Schutz vor weiterer Zerstörung gegeben ist. Besonders gefährdet sind Werkstücke aus unedlen Metallen, wenn diese edlere Metalle berühren (Messingschrauben in Aluminium; verchromtes Eisen mit verletzter Chromschicht), wobei schon eine kleine Verunreinigung der Oberfläche mit einem edleren Metall zur Bildung eines Lokalelementes führen kann.
Bewusst wird der beschriebene Effekt bei Opferanoden eingesetzt. Hier wird das vor Korrosion zu schützende Metall (zum Beispiel Pipelines, Schiffe) leitend mit massiven Stücken aus unedlen Metallen wie Aluminium, Magnesium oder Zink verbunden. Der Oxidation fällt nur das unedle Metall zum Opfer, das Werkstück selbst ist geschützt.
Auch wenn Argon, Kupfer, Diamant und Natriumchlorid hinsichtlich der chemischen Bindung zu völlig unterschiedlichen Stoffklassen zählen, so lässt doch ihr atomarer Aufbau – ihre Struktur – zahlreiche Gemeinsamkeiten erkennen. Vor allem die Vorliebe der Natur zur Ausbildung der höchstsymmetrischen Varianten der dichtesten Kugelpackungen schafft viel Ordnung in der Kristallchemie.
In den chemisch-präparativen Fortgeschrittenenpraktika werden Sie die Unverträglichkeit von Flüssig-Stickstoff-gekühlten „Kühlfallen“ und Argon-Schutzgas kennenlernen: Argon erstarrt bei dieser Temperatur und man kann schnell größere Mengen dieser schneeartigen Substanz ausfrieren. Die niedrigen Phasenumwandlungstemperaturen (Siedepunkt: −185,9 °C, Schmelzpunkt: −189,4 °C) von Argon deuten auf sehr schwache Bindungskräfte zwischen den Argonatomen hin. Es handelt sich um Dispersions- oder van-der-Waals-Wechselwirkungen, die auf der Anziehung induzierter Dipole beruht. Dispersionswechselwirkungen überlagern alle Arten der chemischen Bindung, sie sind aufgrund ihrer äußerst geringen Stärke aber nur dann bemerkbar, wenn andere Wechselwirkungen fehlen. Trotz der geringen Kräfte zwischen zwei Atomen oder kleinen Molekülen bestimmen van-der-Waals-Bindungen die Eigenschaften von Stoffen, wenn deren Bausteine eine ausgedehnte Oberfläche aufweisen, die zahlreiche Berührungspunkte zu Nachbarmolekülen zulässt. Durch Addition vieler kleiner Einzelbeiträge ergibt sich dann eine nennenswerte Wechselwirkung. Lehrbuchbeispiele sind die verschiedenen Alkane, deren Siedepunkt mit der Kettenlänge wächst, bis bei Stoffen wie Polyethylen oder Polystyrol so starke intermolekulare Wechselwirkungen auftreten, dass man diese Kunststoffe als fest empfindet.
Das Umgekehrte gilt, wenn anstelle von Argon ein leichteres Edelgas betrachtet wird. So schmilzt festes Neon bereits bei −248,6 °C, um bei −246,1 °C zu sieden. Helium, das leichteste Edelgas, siedet knapp oberhalb des absoluten Nullpunktes bei −268,9 °C. Als einziges Element wird Helium bei Normaldruck nicht fest. Kristallines Helium kann bei Temperaturen von ca. 1 K untersucht werden, wenn flüssiges Helium unter Druck verfestigt worden ist.
Für die Struktur der Edelgase ist nach dieser Diskussion eine Voraussage möglich: (1) die Atome sollten möglichst dicht gepackt sein, (2) die Umgebung der Atome ist nicht durch gerichtete Bindungen bestimmt. Wie nun sieht eine dichteste Packung gleichgroßer Kugeln aus?
Um eine dichteste Kugelpackung bequem beschreiben zu können, wird zuerst der Begriff „Elementarzelle“ eingeführt. Das Konzept kann angewandt werden, wenn eine (im Vergleich zu atomaren Dimensionen unendlich ausgedehnte) Kristallstruktur – allgemeiner: wenn irgendein Muster – auf einem regelmäßig wiederkehrenden Grundmotiv beruht. Das Muster besitzt in diesem Fall Translationssymmetrie. Ein solches Muster ist dadurch gekennzeichnet, dass der Raum in gleichartig gefüllte Volumina zerteilt werden kann, und zwar so, dass sich aus dem gewählten Raumausschnitt das gesamte Muster wiederherstellen lässt, indem der Ausschnitt entlang seiner Basisvektoren vervielfältigt wird. Das folgende Bild zeigt dieses Prinzip an einem zweidimensionalen Muster:
Zur Aufteilung der Fläche wird ein beliebiger Punkt als Referenzpunkt markiert. Anschließend werden sämtliche Punkte markiert. die exakt dieselbe Umgebung haben. Die so erhaltenen Gitterpunkte bilden nun ein Punktgitter oder kurz Gitter. Die Gitterpunkte werden nun auf zweckmäßige Weise (das heißt so, dass nicht unnötig viele sehr spitze oder sehr stumpfe Winkel auftreten) durch Linien verbunden, welche die Fläche in gleichartige Parallelogramme zerteilt (machen Sie bei der Suche nach einer alternativen Methode mit). Ein solches Parallelogramm heißt Elementarzelle. Ist umgekehrt eine Elementarzelle bekannt, kann das gesamte Muster wiederhergestellt werden. Im Beispiel ist die Elementarzelle ein Rechteck, allgemein ist sie im zweidimensionalen Fall ein Parallelogramm. Darüber hinaus ist die Zelle im Beispielmuster die kleinst mögliche Elementarzelle – sie enthält genau 1 Gitterpunkt (4 × ¼), sie ist primitiv. In drei Dimensionen werden Elementarzellen in der Regel durch das dreidimensionale Analog des Parallelogramms beschrieben, einem Parallelepiped. Auch in dreidimensionalen Zellen enthält die kleinstmögliche, primitive Zelle 1 Gitterpunkt (8 × 1/8).
Nun zu den dichtesten Kugelpackungen. Eine solche Packung kann makroskopisch aufgebaut werden, wenn gleichgroße Kugeln zuerst in der Fläche möglichst dicht gepackt werden: jede Kugel hat sechs nächste Nachbarn, zwischen den Kugeln sind gleichartige Lücken entstanden, die von jeweils drei Kugeln gebildet werden. In der Abbildung wurde der Referenzpunkt für eine Gitterpunktschar in die Mitte einer solchen Lücke gelegt und zwar in eine solche, die unterhalb des Punktes 1 Kugel und oberhalb 2 Kugeln „sieht“. Es lässt sich nun eine Elementarzelle konstruieren, die im Bild rot umrandet ist (das zweite Bild zeigt Ihnen, dass unser Thema auch in der belebten Natur eine Rolle spielt):

Eine Eiablage der Grünen Stinkwanze (Palomena prasina) auf der Unterseite eines Blattes von Strauchbasilikum (Ocimum spec.).
Um nun zu einer dichtesten Kugelpackung in drei Dimensionen zu gelangen, werden in einem ersten Schritt zwei solcher Schichten auf Lücke gestapelt. Schaut man entlang der Packungsnormalen, so findet man nun die Hälfte der Lücken der unteren Schicht von Atomen der oberen Schicht bedeckt, während die andere Hälfte auf Deckung mit Lücken der zweiten Schicht liegt – hier kann man durch die Doppelschicht hindurchsehen:

Als nächstes müsste nun eine dritte Schicht auf die zweite gelegt werden. Da dies graphisch zunehmend unschöner wird, wechseln wir zuvor die Art der Darstellung. Auch wenn wir uns nach wie vor vorstellen, dass die zu packenden Kugeln einander berühren, so stellen wir sie doch in einer Schwerpunktsdarstellung dar, die auch bei den im folgenden präsentierten Kristallstrukturen eine Rolle spielt. Die letzten beiden Bilder sehen nun so aus:

Im unteren Bild ist neben der rot umrandeten Zelle eine alternative primitive Elementarzelle grau eingezeichnet, ferner die übliche Art, die Beträge der Basisvektoren einzuzeichnen. Die Vektoren selbst werden gewöhnlich mit a, b und c bezeichnet, oder, wenn symmetrisch äquivalente Richtungen vorliegen, auch mit a1, a2 und c (2 äquivalente Vektoren) oder a1, a2 und a3 (3 äquivalente Vektoren). Die Beträge der Vektoren und die Winkel zwischen den Vektoren sind die Gitterkonstanten a, b, c, α (Winkel zwischen b und c), β (Winkel zwischen a und c) und γ (Winkel zwischen a und b). Bezeichnungen wie a1 und α1 werden sinngemäß verwendet.
Es gibt nun zwei Möglichkeiten, die erste Kugel einer dritten Schicht auf eine Lücke der zweiten zu legen: entweder auf ein Kugel-Lücke-Paar der unteren Schichten oder auf ein Lücke-Lücke-Paar. Wird die Stapelfolge der beiden unteren Schichten als A und B bezeichnet, so führt die erste Variante zur Stapelfolge ABA. Es ergibt sich wegen der deckungsgleichen Anordnung der beiden A-Schichten eine Darstellung, die von der letzten Graphik nicht zu unterscheiden ist. Die zweite Möglichkeit führt zur Schichtfolge ABC:
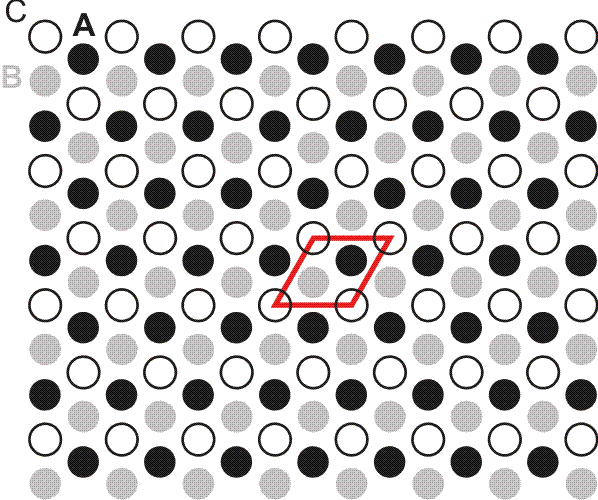
Wie wird die Stapelung fortgesetzt? Durch die Forderung nach einer Stapelung auf Lücke lässt sich ABA gemäß ABAB oder ABAC fortführen, ABC durch ABCA oder ABCB. Beim weiteren Stapeln einer fünften, sechsten, usw. Schicht haben alle denkbaren Varianten dieselbe Raumerfüllung, sie scheinen außerdem energetisch gleichwertig. Der optimistische Gedanke, es könnte in der Natur anstelle einer statistischen Stapelfolge der Art ABCABABACBACACABCB… zu wiederkehrenden, geordneten Varianten der Art (AB)n und (ABC)n kommen, kann daher nicht durch Stabilitätsbetrachtungen belegt werden. Da auf der Basis der Packungsdichte nicht weiter zwischen Stapelvarianten unterschieden werden kann, sollte eine weitere Klärung experimentell erfolgen.
Die einfachste denkbare Schichtfolge ABABAB… ≡ (AB)n, die eine Ordnung in der dritten Dimension – der Stapelrichtung – aufweist, hat keine höhere Raumerfüllung als jede andere Stapelfolge. Es sollte daher nicht erwartet werden, dass sie in der Natur eine Rolle spielt – doch das Gegenteil ist der Fall. Die sogenannte hexagonal dichteste Kugelpackung (hdP, oft auch hcp von engl. hexagonal closest packing) ist die häufigste Elementstruktur im Periodensystem. Mehr als 1/3 aller Elemente kristallisiert in diesem Strukturtyp.
Unter den Edelgasen ist es das unter Druck kristallisierte Helium, das in dieser Struktur gefunden wird. Das obere Bild zeigt den Aufbau eines Heliumkristalls unter Berücksichtigung des van-der-Waals-Radius von Heliumatomen. Die Deutung von van-der-Waals-Radien gelingt mit einem Bild, dass bei der Diskussion der Ionenbindung bereits verwendet wurde: Nähert man zwei Atome einander an, so wird eine anziehende Kraft wirksam – hier eben Dispersionswechselwirkungen – bis der Abstand der Atome so klein ist, dass eine mit abnehmendem Abstand schnell ansteigende Abstoßung der Elektronenhüllen einsetzt. Die Summe aus beiden Wechselwirkungen ergibt ein Energieminimum, das bei einer van-der-Waals-Wechselwirkung den Gleichgewichtsabstand der Atome beschreibt. Der van-der-Waals-Radius ist die Hälfte dieses Abstands.
Die hexagonal dichteste Kugelpackung (gezeichnet mit den Parametern von Helium). Das ideale c/a-Verhältnis ist 1,633, He (ca. 1K, ca. 70 bar): 1,61.
Zur Definition des c/a-Verhältnisses bei der hexagonal dichtesten Kugelpackung.
Die experimentell ermittelte Struktur des Heliums entspricht völlig der Kugelpackung, die aus geometrischen Überlegungen abgeleitet wurde. Dies gilt auch für das Verhältnis von Schichtabstand zu Kugelabstand, für den sich ein Idealwert von 0,8165 für auf Lücke liegende Schichten errechnet. Da in der hdP zwei Schichten pro Elementarzelle vorliegen, ist das ideale c/a-Verhältnis 2 × 0,8165 = 1,6330 (siehe Abbildung).
Die überraschende Vorliebe der Natur für hochsymmetrische Strukturen zeigt sich noch deutlicher, wenn die Struktur der übrigen Edelgase ermittelt wird. Die Abbildung zeigt die Struktur des Argons, dessen van-der-Waals-Radius erwartungsgemäß deutlich größer als derjenige des Heliums ist. Argon kristallisiert in der kubisch dichtesten Kugelpackung (kdP, auch ccp von engl. cubic closest packing oder fcc von engl. face-centered cubic [packing]). Eine nähere Betrachtung zeigt, dass die kdP der Schichtfolge (ABC)n entspricht. Auch die kdP ist sehr häufig; etwas weniger als 1/3 aller Elemente kristallisieren in diesem Strukturtyp. Die erwarteten Stapelvarianten spielen in der Natur eine nur untergeordnete Rolle. Die (unbefriedigend verstandene) Bevorzugung der beiden höchstsymmetrischen Varianten durch die Natur ist ein Ausdruck des Symmetrieprinzips, des Bestrebens der Natur zur Ausbildung möglichst hochsymmetrischer Strukturen.
Die Abbildungen zeigen die Argonstruktur mit van-der-Waals-Radien, als Schwerpunktsmodell und in einer bewegten Darstellung:
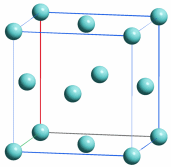
Der Zusammenhang mit der ABC-Schichtfolge wird durch die folgende Bilderserie deutlich. Das erste Bild ist entlang der Stapelachse projiziert, so dass der Blick auf eine dichtest gepackte Schichten fällt (oberste Schicht: 1 Atom, nächst untere Schicht: 6 Atome, nächst untere Schicht: 6 Atome, 4. Schicht verdeckt unter der ersten):
Es wird nun um 10° so um eine waagrecht liegende Achse gedreht, dass sich der obere Teil der Zelle nach hinten bewegt:
… und noch einmal 10°:
… bis der Würfel nach den letzten 10° auf einer Fläche steht:
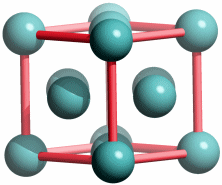
In der letzten Projektion sind weitere Stapelrichtungen zu sehen. Die hohe Symmetrie des kubischen Kristallsystems wird deutlich: obwohl gedanklich mit nur einer Stapelrichtung aufgebaut, weist die Struktur vier gleichwertige Stapelachsen auf – jeweils eine entlang jeder Raumdiagonale des Elementarwürfels.
Auf die große Verbreitung der beiden hochsymmetrischen dichtesten Kugelpackungen unter den Elementstrukturen war hingewiesen worden. Die beiden Packungen sind damit nicht nur die bevorzugte Atomanordnung bei den Edelgasen sondern vor allen Dingen sind sie typische Metallstrukturen. Gilt die Grundvoraussetzung – die ungerichteten Bindungen – für Metalle noch?
¾ aller chemischen Elemente sind Metalle, für welche die folgenden Eigenschaften typisch sind: Die Absorption für sichtbares Licht ist hoch — hieraus folgt das große Spiegelreflexionsvermögen, das die Ursache für Metallglanz und Undurchsichtigkeit ist; hoch sind auch elektrische und Wärmeleitfähigkeit, außerdem sind Metalle verformbar, sie sind duktil. Diese Eigenschaften unterscheiden die Metalle von Stoffen mit ionischer Bindung zwischen den Atomen (Salze) oder mit Atombindungen, die bei den Nichtmetallen häufig sind.
Versuch 28.9: Blattgold und Goldsol projizieren
Versuch 28.10: Kupferstab biegen
In der Nähe der etwa zwischen den Elementen Bor und Tellur verlaufenden Grenze zwischen Metallen und Nichtmetallen liegen die Halbmetalle, die zwar Metallglanz zeigen, sich aber von den Metallen durch die Größe und die Temperaturabhängigkeit der Leitfähigkeit unterscheiden: Elemente wie B, Si, Ge und Te sind Halbleiter mit Leitfähigkeiten, die unter denjenigen der Metalle liegen. Diese geringere Leitfähigkeit steigt mit der Temperatur an, während die Leitfähigkeit eines Metalls mit der Temperatur abnimmt. Besonders im Bereich der Grenzlinie zwischen Metallen und Nichtmetallen ist die Regel zu erkennen, dass der metallische Charakter in einer Gruppe des PSE von oben nach unten und in einer Periode von rechts nach links zunimmt; die Nebengruppenelemente sind sämtlich Metalle.
Die genannten Metalleigenschaften sind an größere Metallatom-Aggregate gebunden: Auch Flüssigkeiten können Metalle sein (Quecksilber; Gallium oberhalb 30 °C), nicht aber Gase, in denen keine größeren Aggregate zu erwarten sind.
Die typischen Metalleigenschaften gehen mit einer charakteristischen Elektronenanordnung bei den Metallen einher, die am Beispiel von Natrium erläutert werden soll: Natrium hat als Element der 1. Hauptgruppe 1 Valenzelektron. Für die Bindung zwischen Natriumatomen scheint weder die Ionenbindung (Na+Na− wird an dem ungünstigen Wert für die Elektronenaffinität von Na scheitern) noch die Atombindung (Na–Na ergibt keine Edelgasschale) eine befriedigende Lösung zu sein. Die hohe elektrische Leitfähigkeit zeigt die Existenz von frei beweglichen Ladungsträgern an; es lässt sich zeigen, dass es sich um Elektronen handelt. Jedes Natriumatom trägt offenbar sein Valenzelektron zu einem Elektronengas bei, in das Na+-Ionen eingebettet sind. Die Bindung zwischen den Na-Atomen ergibt sich dann als elektrostatische Anziehung zwischen den Na+-Ionen und der negativen Ladungsdichte zwischen den Atomrümpfen.
Bei dieser Betrachtungsweise entspricht das Prinzip der Metallbindung dem der Atombindung (vergleiche die Elektronendichteverteilungen zwischen den Atomkernen bei den verschiedenen Bindungsarten). Der Unterschied zwischen Metall- und Atombindung besteht in der räumlichen Ausrichtung der Bindungselektronen bei der Atombindung. Die Metallbindung ist dagegen ungerichtet, wodurch die wenigen zur Verfügung stehenden Elektronen mit mehr Atomrümpfen wechselwirken können (vergleiche: in CO2 werden drei Atome durch acht Elektronen verbunden, in einem Natriumkristall werden die acht nächsten Nachbarn eines jeden Na-Atoms durch zwei Elektronen gebunden).
Versuch neu: Abhängigkeit der Leitfähigkeit von der Temperatur bei Metallen und Halbleitern
Die Strukturbestimmung an sehr verschiedenen, aber wichtigen Metallen wie dem reaktiven Natrium, dem zähen Hartmetall Wolfram und dem wichtigen Gebrauchsmetall Eisen führt zu einem völlig unerwarteten Ergebnis: keines dieser Elemente wählt eine der beiden dichtesten Kugelpackungen noch Stapelvarianten als Struktur. Sie sind vielmehr auf eine Art und Weise aufgebaut, die bei der makroskopischen Betrachtung zur Kugelpackung überhaupt nicht erfasst wurde – der kubisch innenzentrierten oder kubisch raumzentrierten Kugelpackung (engl. bcc von body-centered cubic). Die Elementarzelle ist denkbar einfach: in einem Würfel sind die Eckpunkte und die Raummitte durch Atome besetzt. Diese sind alle auch bezüglich der Orientierung der nächsten Nachbarn äquivalent (sie sind translatorisch äquivalent), daher enthält die Elementarzelle auch zwei Gitterpunkte. Der zweite Gitterpunkt in der Zellmitte (nicht das Atom!) führt zum Begriff „innen- oder raumzentriert“.
Die Packung weist durchaus Beziehungen zu den dichtesten Kugelpackungen auf. Das folgende Bild zeigt den Blick auf eine Ebene, für welche die Elementarzelle entlang einer Diagonale aufgeschnitten wurde. Der Bezug zur dichtesten Packung ist offensichtlich: von den sechs nächsten Nachbarn innerhalb einer Schicht wird die mittlere Kugel nur noch von vieren berührt, während die beiden übrigen den unmittelbaren Kontakt verloren haben, aber trotzdem nicht weit entfernt sind:
In drei Dimensionen betrachtet, zeigen sich weitere Charakteristika. Die mittlere Kugel hat 8 unmittelbare Nachbarn, weitere 6 Nachbarn – die Mittelpunkte der sechs Nachbarzellen – sind jedoch nur wenig weiter entfernt. Ist der Abstand zur nächsten Kugel 1, dann ergibt sich für den Abstand zur übernächsten Kugel 2/√3 = 1,155, die 6 übernächsten Nachbarn sind also nur um ca. 15 % weiter entfernt als die 8 direkten Nachbarn:
Können Sie den Abstand der übernächsten Nachbarn in der kdP angeben, wenn der Abstand der nächsten Nachbarn 1 ist?
Noch eine Anmerkung zur Elementarzelle der kubisch-raumzentrierten Packung: wenn Ihnen die Abbildungen alle zu klein sind, um sich die Struktur anschaulich vorstellen zu können – besuchen Sie Brüssel. Dort finden Sie eine Vergrößerung:

Das Atomium in Brüssel, hier während der Anfang 2006 abgeschlossenen Renovierung, zeigt die kubisch-raumzentrierte Elementarzelle von α-Eisen in 165-milliardenfacher Vergrößerung als Schwerpunktmodell (Foto: B. Leiter).
Beim Aufbau der dichtesten Kugelpackungen war aufgefallen, dass regelmäßig verteilte Lücken zwischen den Kugeln auftreten. Der Anteil des Raums, der in einer Kugelpackung von Kugeln eingenommen wird, ist die Raumerfüllung. Sie wird meist in Prozent angegeben. Für die kubischen Strukturen lässt sich die Raumerfüllung leicht berechnen.
Die kdP enthält vier Kugeln pro Elementarzelle. (Da die Umgebung aller Kugeln identisch ist, enthält die Zelle auch 4 Gitterpunkte – es sollte also möglich sein, eine primitive Zelle in das Gitter einzuzeichnen, die nur ¼ so groß ist wie die abgebildete Standardzelle. Können Sie eine solche primitive Zelle einzeichnen?) Die Kugeln berühren einander entlang der Flächendiagonalen; dabei enthält jede Flächendiagonale 4 Atomradien (4 r). Welches Volumen nehmen diese vier Kugeln ein? Wenn r als 1 definiert wird, ist das gesuchte Volumen VK das 4-fache des Volumens einer einzelnen Kugel:
VK = 4 · 4/3 π r3
Die Raumerfüllung wird erhalten, wenn VK durch das Volumen V = a3 der Elementarzelle mit der Gitterkonstanten a dividiert wird. Um a zu ermitteln, wird die Beziehung
√2 a = 4 r
verwendet. Mit dem Elementarzellvolumen
V = (4 r/√2)3 = 16 √2 r3
ergibt sich die gesuchte Raumerfüllung zu
| VK | = | 16/3 π r3 | = | π | = 0,7405 |
| V | 16 √2 r3 | 3 √2 |
In den dichtesten Kugelpackungen sind also 74 % des Raumes mit Kugeln gefüllt, 26 % fallen auf die Lücken.
Wie ist die Raumerfüllung bei der kubisch innenzentrierten Packung? Hier enthält die Elementarzelle 2 Kugeln (und 2 Gitterpunkte), also:
VK = 2 · 4/3 π r3
Die Abbildung der Struktur zeigt, dass nun gilt:
√3 a = 4 r
und weiter
V = (4 r/√3)3 = 64/(3 √3) r3
Die Raumerfüllung ist nun:
| VK | = | 8/3 π r3 | = | √3 π | = 0,6802 |
| V | 64/(3 √3) r3 | 8 |
Mit 68 % Raumerfüllung ist die kubisch innenzentrierte Packung tatsächlich keine dichteste Kugelpackung. Sie wurde daher auch nicht bei der Suche nach dichtesten Packungen erkannt.
Kugelpackungen sind in vielen Disziplinen von Interesse, entsprechend groß ist die Fülle an Begriffen rund diese Packungen. Hier nur eine Auswahl:
Packungen aus gleichartigen Kugeln in symmetrisch äquivalenter Umgebung sind homogene Kugelpackungen. Diese sind neben Parametern wie der Schichtfolge auch dadurch charakterisiert, wieviel berührende Nachbarn jede Kugel aufweist. So sind hdP und kdP 12-er Kugelpackungen, während die kubisch innenzentrierte Packung eine 8-er Kugelpackung ist. Je kleiner die Zahl der nächsten Nachbarn, umso geringer die Packungsdichte der homogenen Kugelpackung. Die Metallurgie hat weitere Bezeichnungen beigesteuert: die kdP ist dort als A1-Typ, die hdp als A3-Typ und die innenzentrierte Packung als A2-Typ bekannt. In der Kristallchemie schließlich sind der A1-, A2- und A3-Typ der Kupfer-, Wolfram- und Magnesiumtyp, meist kurz: Cu-Typ, W-Typ und Mg-Typ. Die Tabelle fasst zusammen:
| Cu-Typ | W-Typ | Mg-Typ | |
|---|---|---|---|
| englisch (deutsch) | fcc (kdP) | bcc (krz) | hcp (hdP) |
| Metallurgie | A1 | A2 | A3 |
| homogene … Packung | 12-er | 8-er | 12-er |
| Raumerfüllung | 0,74 | 0,68 | 0,74 |
| nächste Nachbarn | 12 | 8 + 6 | 12 |
| Gitterpunkte pro EZ | 4 | 2 | 1 |
| Kugeln pro EZ | 4 | 2 | 2 |
Ca. 80 % der metallischen Elemente kristallisieren in den Strukturtypen A1, A2 und A3:
• A1-Typ: Cu, Ag, Au, Pt, γ-Fe, Al, Pb, Ni und andere
• A2-Typ: Alkalimetalle, Ba, V, Cr, Mo, W, α-Fe
• A3-Typ: Mg, Ti, Co
Viele Metalle sind polymorph, das heißt in Abhängigkeit von der Temperatur kristallisieren sie in mehr als einer Struktur. Eisen ist das wichtigste Beispiel aus der Technik:
Eisen ist in der γ-Form duktiler und damit leichter bearbeitbar als in der α-Form. Diese Abhängigkeit der Verformbarkeit von der Kristallstruktur zeigt, dass die für die Duktilität verantwortliche Verschiebbarkeit entlang Ebenen im Innern der Kristalle in den einzelnen Strukturtypen verschieden ist. Hohe Duktilität wird begünstigt, wenn in den Kristalliten der polykristallinen Metallstücke zum einen die Gleitebenen möglichst glatt sind – dies ist gleichbedeutend mit einer möglichst dichten Packung auf der Ebene, und wenn zum anderen glatte Ebenen in möglichst unterschiedlichen Orientierungen auftreten, was die Wahrscheinlichkeit erhöht, dass geeignete Gleitebenen sich in einer günstigen Orientierung zur verformenden Kraft befinden.
Im A1-Typ sind beide Forderungen am besten erfüllt: Es treten dichtest gepackte Schichten auf; diese verlaufen in vier Orientierungen durch die kubische Elementarzelle (senkrecht zu den vier Raumdiagonalen des Würfels). Im A3-Typ kommen dichtest gepackte Ebenen vor, aber nur in einer Orientierung, nämlich senkrecht zur Stapelrichtung eben dieser Ebenen; weitere optimale Gleitebenen ergeben sich durch die Symmetrie der Struktur nicht, wie dies im kubischen Kristallsystem der Fall ist. Der A2-Typ ist wie der A1-Typ kubisch, so dass alle betrachteten Ebenen durch die hohe Symmetrie in verschiedenen Raumrichtungen erscheinen, aber es gibt keine so dicht gepackten Ebenen wie in den beiden Strukturen mit dichtester Kugelpackung. Die Ebenen sind „rauher“, da die Ebenen mit 6-er-Koordination (das sind die durch eine Würfelkante und eine Flächendiagonale definierten Ebenen) nicht dichtest gepackt sind. Entsprechend diesen Struktureigenheiten sind A1-Metalle duktiler als die meist spröderen Metalle mit A2- und A3-Struktur.
Die Sprödigkeit vieler Metalle erhöht sich, wenn nichtmetallische Verunreinigungen wie Sauerstoff, Stickstoff oder Kohlenstoff in das Metallgitter eingebaut sind. Gerade die in diesem Bindungszustand kleinen Kohlenstoffatome, die ohne weitere Änderung der Metallstruktur einige Lücken des Gitters besetzen, führen zu drastisch verminderter Duktilität, es entstehen Hartstoffe.
Die Natriumchlorid-Struktur ist bisher nur aus dem Blickwinkel der Koordinationspolyeder betrachtet worden. Wie aber lässt sich die Gesamtstruktur einordnen? Eine Vorhersage scheint schwierig: die Bindungen sind zwar nicht gerichtet, dafür aber sind die Kuglen verschieden groß und es gibt die Randbedingung, dass jede Kugelsorte nur Kugeln der anderen Sorte anzieht, gleichnamige Kugeln aber abstößt. Das Experiment ergibt die oben bereits gezeigte Struktur:
Versuch 12-6: Salzstrukturen
Die Beziehung zur kdP ist offensichtlich: Die größeren Ionen (Cl−) bilden eine kdP. Diese ist aufgeweitet, wodurch Raum für die Natrium-Ionen entsteht. Die kleineren Kationen haben den Raum aufgeweitet, der in der kdP dadurch entstanden war, dass eine Lücke zwischen drei Kugeln auf Deckung mit einer anderen Lücke zu liegen kam. Wegen der oktaedrische Umgebung der Lücke mit sechs Atomen der dichtesten Packung heißt sie Oktaederlücke. In einer Packung aus n Kugeln bilden sich n Oktaederlücken, die in der NaCl-Struktur alle besetzt sind: NaCl ist eine AB-Verbindung (nicht mit irgendeiner Schichtenfolge verwechseln!)
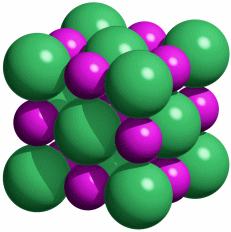
Es ist sinnvoll, die größere Ionensorte als packungsbildend anzusehen und den kleineren Ionen die Lücken zuzuweisen. Die Beobachtung, dass die Anionen aber nicht mehr dichtest gepackt sind, sondern dass die Anion-Teilstruktur aufgeweitet ist, zeigt schon, dass man das Konzept der gefüllten Lücken nicht zu wörtlich nehmen sollte. Dies wird im folgenden Bild noch deutlicher:
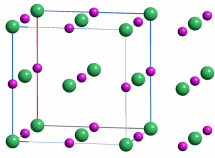
Wird der Ursprung der Elementarzelle um ½ nach rechts verschoben, wird eine gleichwertige Zelle erhalten, bei der nun die Positionen von Anionen und Kationen gegenüber der ersten Zelle vertauscht sind. Formal bilden nun die kleineren Kationen eine kdP, in deren Oktaederlücken sich die größeren Anionen befinden. Das Beispiel lehrt: nehmen die Kugeln einer Teilstruktur die Plätze der Kugeln einer dichtesten Packung ein, so belegt dies noch nicht, dass im Sinne einer wirklich dichtesten Packung Kontakt zwischen den Kugeln besteht; es ist elektrostatisch keineswegs ungünstig, wenn gleichnamige Ionen durch Gegenionen – auch weit – auseinandergeschoben werden, wenn die Gegenionen zu groß sind, um sich in eine echte dichteste Packung einzufügen. Das Prinzip der dichtesten Packung liefert vielmehr erst dann ein Stabilitätsargument, wenn die kleinere Ionensorte zu klein ist, um die Lücken in der Packung der größeren auszufüllen. Dieser Fall wird weiter unten behandelt.
Bevor weitere Ionenstrukturen behandelt werden, kann bereits jetzt die Frage nach der Sprödigkeit von Salzkristallen beantwortet werden. Die kubisch dichteste Kugelpackung war bei den Metallen stets mit besonders hoher Duktilität verbunden, trotzdem sind NaCl-Kristalle spröde. Der Grund liegt im Aufbau des Salzes aus Anionen und Kationen. Wird durch eine scherende Kraft ein Teil des Kristalls über den anderen hinweg bewegt, dann kommt es zwar zu einem Gleiten von Atomen über glatte Flächen, dies führt aber dazu, dass nach einer halben Gitterkonstante gleichnamige Ionen in engen Kontakt kommen – der Kristall bricht an der Scherebene auseinander.
Nicht alle Salze mit AB-Zusammensetzung kristallisieren in der NaCl-Struktur. So wird für Caesiumchlorid der folgende Aufbau gefunden (Cs violett, Cl grün):
Die primitive(!) kubische Zelle enthält nun 1 Cs-Ion und 8/8 Chlorid-Ionen – eine AB-Struktur eben. Wie bei der NaCl-Struktur sind die Kation- und die Anion-Teilstruktur translatorisch gleichwertig:
Jedes Ion ist würfelförmig von 8 Gegenionen umgeben, die Koordinationszahl ist also größer als bei der NaCl-Struktur mit 6. Wozu braucht die Natur zwei Strukturtypen für so ähnliche Ionenverbindungen? Betrachten Sie hierzu das folgende Bild, bei dem die vorderen zwei der acht Chlorid-Ionen weggelassen sind:
Der typische Aufbau eines Ionenkristalls wird deutlich: Anionen und Kationen berühren einander, die gleichgeladenen Ionen haben dagegen einen größeren Abstand voneinander. Den anziehenden Cs-Cl-Wechselwirkungen stehen abstoßende Cs-Cs- und Cl-Cl-Wechselwirkungen gegenüber. Lassen Sie nun in Gedanken das Kation schrumpfen. Die Anionen werden dem Kation folgen um den Kontakt aufrecht zu erhalten, die Elementarzelle wird kleiner, die Anionen rücken entlang der Basisvektoren näher aufeinander zu. Schließlich kommt es zum Anion-Anion-Kontakt. Schrumpft nun das Kation weiter, würde ein möglicher kürzerer Kation-Anion-Kontakt verschenkt. Unterhalb dieses Grenzradius, bei dem Kontakt gleichnamiger Ionen auftritt, ist es für die Natur günstiger, auf eine Struktur mit kleinerer Koordinationszahl (kleineren Lücken) umzuschalten, um anziehende Wechselwirkungen hinzu zu gewinnen. Machen Sie sich bei der ganzen Diskussion klar, dass das Coulombsche Gesetz gilt: in dieses gehen keine Ionenradien ein, sondern nur die Lage der Ladungsschwerpunkte. Der Coulomb-Anteil der Gitterenergie ändert sich nicht, gleichgültig ob das Kation seine Lücke ausfüllt oder nicht. Die höher koordinierte Struktur kollabiert bei zu kleinem Gegenion nicht weil sie unstabil wird, sondern weil es eine stabilere Alternative gibt.
Für eine Ionenverbindung ist das Grenzradienverhätnis eine charakteristische Größe. Nachdem deutlich wurde, dass bei diesem kleinst möglichen Radienverhältnis Kontakt der größeren Ionensorte vorliegt, können einzelnen Ionen Radien zugeordnet werden. Machen Sie sich klar, dass allein aus Kation-Anion-Abständen keine Ionenradientabelle abgeleitet werden kann, da die Rechnung unterbestimmt ist. Um individuellen Ionen einen Radius zuweisen zu können, muss ein Radius irgendeines Ions bekannt sein, um anschließend die übrigen Kation-Anion-Abstände in zwei Beiträge zerteilen zu können.
Eine der bekanntesten und vollständigsten Radientabellen wurde von Shannon und Prewitt vorgelegt. Die folgende Tabelle enthält unkorrigierte Shannon-Radien, darunter werden Radien verstanden, die auf der ursprünglichen Festlegung beruhen, für das Oxid-Ion bei der Koordinationszahl 6 einen Wert von 126 pm anzugeben. In der Literatur ebenfalls verbreitet sind korrigierte Shannon-Radien, bei denen alle Anionradien um 16 pm vergrößert und alle Kationradien um 16 pm verkleinert wurden. Der Sinn ist, die neuere Shannon-Tabelle mit der älteren Pauling-Tabelle in Einklang zu bringen. Ein beliebter Fehler: Werte aus beiden Tabellen zu mischen; um Ionenradien aus verschiedenen Tabellen korrekt zu kombinieren, muss bei allen verwendeten Tabellen der Ionenradius des Oxid-Ions gleich sein!
Tabelle: Unkorrigierte Shannon-Radien in pm für die Koordinationszahlen 4, 6 und 8.
| 4 | 6 | 8 | |
|---|---|---|---|
| Li | 73 | 90 | 106 |
| Na | 113 | 116 | 132 |
| K | 151 | 152 | 165 |
| Rb | 166 | 175 | |
| Cs | 181 | 188 | |
| Be | 41 | 59 | |
| Mg | 71 | 86 | 103 |
| Ca | 114 | 126 | |
| Sr | 132 | 140 | |
| Ba | 149 | 156 | |
| O | 124 | 126 | 128 |
| S | 170 | ||
| Se | 184 | ||
| Te | 207 | ||
| F | 117 | 119 | |
| Cl | 167 | ||
| Br | 182 | ||
| I | 206 |
Man beachte, dass die Angabe verschiedener Radien für verschiedene Koordinationszahlen mit der Vorstellung, dass sich Ionen wie harte Kugeln verhalten, nicht verträgt. Auf diesen Punkt wird weiter unten eingegangen.
Der kritische kleinste Wert für das Radienverhältnis von kleinerem zu größerem Ion kann in ähnlicher Weise durch eine Betrachtung der Elementarzelle ermittelt werden wie es bei der Bestimmung der Raumerfüllung benutzt wurde. Für die CsCl-Struktur lässt sich der gesuchte Wert ableiten, indem eine Ebene betrachtet wird, die sowohl Kation-Anion- wie auch Anion-Anion-Kontakte enthält. Dies gilt für das Rechteck, das von einem Basisvektor und der zu diesem senkrecht stehenden Flächendiagonale gebildet wird [wenn Sie schon wissen, was Millersche Indizes sind: die (110)-Fläche]. Wenn richtig gerechnet worden ist, kommt heraus: rklein/rgroß = √3 − 1 = 0,732 heraus. Liegt der Radienquotient unter dieser für die CsCl-Struktur kritischen Zahl, so wird an deren Stelle die NaCl-Struktur ausgebildet.
In analoger Weise kann das Grenzradienverhältnis für die NaCl-Struktur berechnet werden. Es beginnt wieder mit der Suche nach einer Fläche, die beide Kontakte enthält – hier jede der sechs Würfelflächen [die (100)-Fläche]. Die Rechnung führt zum Ergebnis: rklein/rgroß = √2 − 1 = 0,414. Liegt das Radienverhältnis unter diesem Wert, so wird anstelle der NaCl-Struktur eine AB-Struktur ausgebildet, in der eine kleinere Koordinationszahl als sechs realisiert ist – Blende- oder Wurtzit-Struktur.
Man beachte, dass die errechneten Grenzradienverhältnisse die Struktur ausschließen, bei welcher die Ionenradienquotient unter dem Grenzwert liegt. Der umgekehrte Schluss gilt nicht, so gibt es zahlreiche NaCl-Typ-Salze mit einem Radienverhältnis im Bereich zwischen 0,732 und 1. Konkret: Alle Alkalimetallhalogenide kristallisieren im NaCl-Typ, nur CsCl, CsBr, und CsI nicht. Der CsCl-Typ ist offensichtlich für eine Ionenverbindung wenig attraktiv – obwohl seine Madelung-Konstante die größte unter den AB-Salzen ist.
| Typ | A |
|---|---|
| CsCl | 1,76267 |
| NaCl | 1,74756 |
| Blende | 1,63805 |
| Wurtzit | 1,64132 |
Wir diskutieren die Ursache für die Schwäche der CsCl-Struktur; eine weitere Frage: wieso wählt CsF den NaCl-Typ?
Mit der Zinkblendestruktur wird der Bereich der Ionenverbindungen endgültig verlassen, der Lückenformalismus ist jetzt nur noch als topologische Hilfe für die Beschreibung einer Kristallstruktur zu gebrauchen. Dies wird deutlich, wenn ein und dieselbe Kristallstruktur mit unterschiedlichen Bindungstypen verknüpft wird. Für Zinkblende, ZnS, gibt die Elektronegativitätsdifferenz Auskunft über den Bindungstyp: Mit χPauling(Zn) = 1,65 und χPauling(S) = 2,58 ergibt sich eine Differenz von ca. 0,9, Zn-S-Bindungen haben also überwiegend kovalenten Charakter. Die Darstellung mit Ionenradien ist daher wenig adäquat (und die Berechnung eines Grenzradienverhältnisses für eine Ionenverbindung mit ZnS-Struktur wenig realistisch):
Wie stark jede Radiendiskussion vom Bindungstyp abhängt, zeigt die Darstellung der Blendestruktur mit kovalenten Radien:
Bei beiden Darstellungen berühren die Kugeln einander, es treten weder Lücken entlang der Bindungslinie auf noch durchdringen die Kugeln einander. Die Summe beider Arten von Radien beschreibt also befriedigend den Abstand der Atome in ZnS. Das Problem besteht vielmehr darin, entlang der Bindung zu entscheiden, wo das eine Atom aufhört und wo das andere anfängt – genau das versucht man bei der Zuweisung eines Radius.
Für eine topologische Beschreibung der Struktur kann man die Bindungsdiskussion umgehen und die Struktur mit Hilfe eines Schwerpunktmodells betrachten:
Dabei wird deutlich, dass die Schwefelatome eine kdP bilden. Die Zinkatome besetzen nun Tetraederlücken, von denen acht in der Elementarzelle der kdP vorliegen. In der Blendestruktur ist demnach die Hälfte der Tetraederlücken besetzt. Nach der Unsicherheit bei der Bindungsbeschreibung wundert es nicht mehr, dass – wie die NaCl-Struktur – auch die Blendestruktur alternativ beschrieben werden kann: Wird der Ursprung der Elementarzelle um ¼ ¼ ¼ verschoben, so bilden nun die Zinkatome eine kdP und die Schwefelatome liegen in der Hälfte der Tetraederlücken. Eine äquivalente Beschreibung ergibt sich, wenn nur die kdP-Teilstrukturen betrachtet werden: Die Blendestruktur besteht demnach aus zwei kubisch dichtesten Kugelpackungen, die um den Vektor ¼ ¼ ¼ gegeneinander verschoben sind (NaCl ist in dieser Betrachtungsweise analog aufgebaut, lediglich der Verschiebungsvektor ist ein anderer, nämlich ½ 0 0). Ist es Zufall, das kovalente Zinksulfid in einer Tetraederstruktur (das ist eine Struktur, in der alle Atome tetraedrisch koordiniert sind) zu finden? Natürlich nicht. Die Ionenbindung war als Bindungstyp eingeführt worden, in dem ungerichtete Bindungen vorherrschen. Das Tetraeder aber ist die typische Koordinationsfigur für kovalente Verbindungen mit einem Valenzelektronen-Oktett. Die Valenzelektronensumme in ZnS beträgt 2 + 6 = 8, im Mittel also 4 Elektronen pro Atom, die aufgrund der geringen Elektronegativitätsdifferenz in vier kovalenten Bindungen gepaart werden können.
Die Wurtzit-Struktur ist das Analog der Blendestruktur, allerdings aufbauend auf einer hdP. Die Zahl und die Art der Lücken bleibt gleich, auch die hdP enthält gleich viele Oktaederlücken wie packungsbildende Atome und doppelt so viele Tetraederlücken.
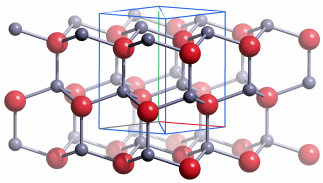
Die Wurtzit-Struktur, gezeichnet mit den Parametern des Zinkits, ZnO.
Auch Wurtzit ist eine Tetraederstruktur, lediglich die Form der sechsgliedrigen Zn3S3-Ringe ist anders: während Blende ausschließlich Sechsringe in Sesselkonformation enthält, liegen beim Wurtzit neben Sechsringen in Sessel- auch solche in Wannenkonformation vor (auch Bootkonformation genannt).
Auch zur Natriumchlorid-Struktur gibt es ein hexagonales Analog: die Nickelarsenid-Struktur, in der beide Atomsorten sechsfach von der jeweils anderen umgeben ist:
Die NiAs-Struktur (Ni: bläulich, As violett; willkürliche Radien). Alle Nickelatome, aber nur die beiden linken Arsenatome haben Anteil an der Elementarzelle. Man beachte die Stauchung der hexagonal dichtesten Packung der Arsenatome entlang c (c/a = 1,39).
Die NiAs-Struktur ist bei Ionenverbindungen außerordentlich selten, sie scheint keine Alternative zur NaCl-Struktur zu sein. Man beachte die Flächenverknüpfung der NiAs6/6-Oktaeder, um den Nachteil gegenüber der NaCl-Struktur mit ihrer Kantenverknüpfung von NaCl6/6-Oktaedern zu erkennen.
Die Betrachtung von Zinkblende als Tetraederstruktur führt natürlich unmittelbar zu Elementstrukturen aus vierbindigen Atomen. Prototyp ist die Diamantstruktur, die entsteht, wenn alle Zink- und Schwefelatome durch Kohlenstoffatome ersetzt werden (bewegtes Bild):
Parameter von Si (a = 545 pm)
Der Aufbau aus C6-Sesseln wird deutlich, ebenso der auffällige Adamantan-Ausschnitt.
Das Paar Wolfram-Typ/Caesiumchlorid-Typ ist sehr ungewöhnlich und es gibt kein analoges Paar bei den dichtesten Kugelpackungen. Wird gedanklich vom W-Typ ausgegangen, so erhält man den CsCl-Typ, indem in geordneter Weise die Hälfte der W-Atome durch Kationen und die andere Hälfte von Anionen substituiert wird. Im Gegensatz zu den übrigen Salzstrukturen werden also nicht Ionen in Lücken einer dichten Packung von Gegenionen eingefüllt, sondern die Salzstruktur entsteht durch Austausch packungsbildender Atome im Verhältnis 1:1. Der Übergang von einer Metallstruktur zu einer Salzstruktur durch bloßen Austausch von Atomen macht W- und CsCl-Typ zum geeigneten Strukturtyp, um Übergänge zwischen Metallen und Salzen zu realisieren. Solche Verbindungen sind immer dann zu erwarten, wenn ein nennenswerter Elektronegativitätsunterschied zwischen den Partnern vorliegt (→ Salz), ohne dass aber der elektronegative Partner ein typisches Nichtmetall ist und somit Elektronenmangel herrscht (→ Metall). Zahlreiche Beispiele stellen daher 1:1-Kombinationen aus einem stark elektropositiven Metall (1. oder 2. Hauptgruppe) und einem Element aus der Mitte des Periodensystems dar, zum Beipiel CaHg (χPauling: Ca = 1,0; Hg 2,0) oder CsAu (χPauling: Cs = 0,8; Au 2,5).
Man kann sich fragen, warum es nicht eine superdicht gepackte Salzstruktur gibt, die durch analoge 1:1-Substitution der Kugeln einer dichtesten Packung entsteht. Haben Sie eine Idee?
Das Paar Diamant-Typ und Blende-Typ bietet einen ähnlichen Übergang, nun aber zwischen rein kovalent gebundenen Stoffen und (beinahe) Ionenverbindungen. Man betrachte im Periodensystem die Elementfolge
Cu Zn Ga Ge As Se Br
Germanium besitzt als Element der 4. Hauptgruppe 4 Valenzelektronen und kristallisiert im Diamant-Typ. Durch Kombination von jeweils zwei Elementen, die gleich weit links und rechts von Ge stehen, werden zunehmend ionischere Verbindungen erhalten, die alle im Blende-Typ kristallisieren:
GaIIIAs−III → ZnIISe−II → CuIBr−I
Verbindungen mit AB2-Stöchiometrie lassen sich ähnlich klassifizieren wie AB-Strukturen. Eine weit auseinandergeschobene kdP von Ca2+-Kationen bildet die Grundlage für eine Interpretation der CaF2-, der Fluorit-Struktur (nicht den Mineralnamen Fluorit mit dem Fluorid-Ion verwechseln! Fluorit ist Calciumfluorid.)
Das Schwerpunktmodell zeigt noch deutlicher, dass die Fluorid-Ionen alle Tetraederlücken der Calciumpackung besetzen.
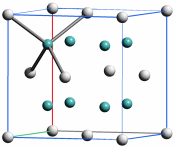
Bei der Stöchiometrie AB2 müssen sich die Koordinationszahlen von A und B wie 2:1 verhalten. Bei Fluorit ist dies 8:4, indem jedes Calcium-Ion würfelförmig von 8 Fluorid-Ionen und jedes Fluorid-Ion tetraedrisch von vier Calcium-Ionen umgeben ist. Auch in der AB2-Reihe wird die hohe Koordinationszahl von 8 für das Kation nur bei einem großen Radienverhältnis gefunden. (Können Sie ausrechnen, dass rA/rB größer als √3 − 1 = 0,732 sein muss?) Ist das Grenzradienverhältnis unterschritten, kommt es zur Ausbildung eines besonders wichtigen und verbreiteten Strukturtyps, der Rutil-Struktur.
Die Rutil-Struktur lässt sich nicht wie die bisher behandelten Strukturen aus einer einfachen Kugelpackung ableiten (wenn Sie so etwas spannend finden: die Oxid-Ionen bilden eine homogene 11-er Kugelpackung). Es ist daher üblich, von der oktaedrischen Koordination der Kationen auszugehen und die Verknüpfung der TiO6/3 zu betrachten. Das Bild zeigt drei Elementarzellen, wodurch hervorgehoben wird, dass die Struktur entlang der senkrechten Achse von kantenverknüpften Oktaeder durchzogen ist. Zwischen diesen Strängen sind die TiO6/3-Oktaeder eckenverknüpft. Die Elementarzelle hat eine andere Form als bisher, Rutil gehört zum tetragonalen Kristallsystem. Titanatome finden sich an den Eckpunkten der Zelle und in der Raummitte. Ist das Gitter deshalb raumzentriert wie im Wolfram-Typ?
Sinken die Koordinationszahlen auf 4:2, liegen so „luftige“ Strukturen vor, dass ihr Zusammenhalt gerichtete Bindungen erfordert. Wichtige Beispiele werden bei den SiO2-Modifikationen gefunden (Modifikationen sind verschiedene Kristallformen bei gleicher Zusammensetzung). Die Abbildung zeigt die Struktur von Quarz (engl. quartz) in zwei Projektionen. Eine nähere Betrachtung zeigt, dass im Quarz (Bergkristall, Seesand) gleichsinnig angeordnete Schrauben vorliegen, Quarz ist chiral (griechisch für „händig“).
Eine zweite SiO2-Modifikation ist Tridymit. Es entsteht ein Eindruck, in welch vielfätiger Weise SiO4/2-Tetraeder in der Lage sind, luftige Raumnetzstrukturen aufzubauen.
Ein besonders interessanter Punkt: wo haben Sie die Tridymit-Struktur schon einmal gesehen?
Atomradien, Metallradien, Ionenradien, kovalente Radien, van-der-Waals-Radien: So nützlich Radien sind, zum Beispiel um Bindungslängen vorherzusagen, so verwirrend sind die verschiedenen Konzepte. Auf der Grundlage der Strukturchemie gelingt jedoch eine Übersicht: alle diese Radien außer den Ionenradien sind vom Grundkonzept her einfach: „Atomradius“ ist eine Sammelbezeichnung; ist das betrachtete Atom ein Metall, so ist damit der halbe Abstand zum nächsten Nachbarn gemeint; oft sagt man dann Metallradius. Ist das betrachtete Atom ein Nichtmetallatom, ist der halbe Abstand zum Bindungspartner in einer kovalenten Bindung gemeint – daher auch die Bezeichnung „Kovalenzradius“. Analog werden van-der-Waals-Radien als halber Abstand zum nächsten Nachbarn definiert, der nicht kovalent angebunden ist. Für Cl2 ergibt sich damit das folgende Bild:
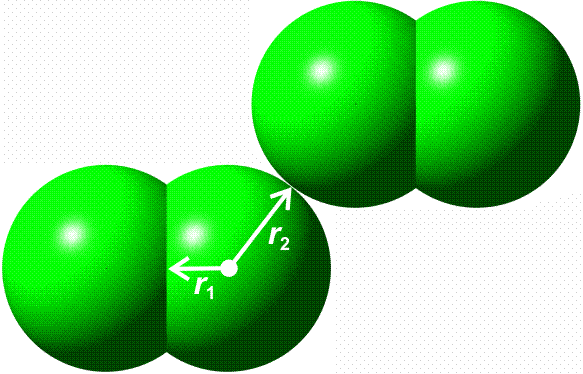
r1 ist der kovalente Radius, r2 der van-der-Waals-Radius. Beide Werte werden der experimentell ermittelten Kristallstruktur des festen Chlors entnommen. Das Problem der Zuordnung von Ionenradien wurde bereits besprochen: Ionenradien sind Radien, die nicht durch Halbierung eines präzise bestimmbaren Abstandes erhalten werden können. Ausgehend von Metallradien sind Kationradien kleiner – wie es zu erwarten ist, wenn weniger Elektronen von der gleichen Kernladung angezogen werden. Anionradien sollten dementsprechend größer als van-der-Waals-Radien sein, sie sind es aber oft nicht. So ist für Chlor:
rvdW(Cl) ≈ rion(Cl−)
Die Ursache ist leicht zu verstehen: van-der-Waals-Kontakte werden durch sehr schwache Bindungskräfte vermittelt, der Gleichgewichtsabstand ist sofort erreicht, wenn eine kleine Abstoßung der Elektronenhüllen spürbar wird. Eine Anion-Teilstruktur ist demgegenüber durch die Wechselwirkung mit der Kation-Teilstruktur massiv komprimiert.
Das Wechselspiel von Kation-Anion-Anziehung und Anion-Anion-/Kation-Kation-Abstoßung führt auch dazu, dass Ionenradien von der Koordinationszahl abhängen: wächst die Koordinationszahl eines Kations, so behindern sich die umgebenden Anionen zunehmend; der Abstand zum Kation wird größer – das Kation scheint größer geworden zu sein.
Ammoniumfluorid, NH4F, ist eine AB-Verbindung aus NH4+- und F−-Ionen. Das Radienverhältnis erlaubt die CsCl-Struktur. Aus den oben besprochenen Gründen kann auch mit der NaCl-Struktur gerechnet werden. Das Ergebnis der Strukturanalyse ist daher erstaunlich: Ammoniumfluorid kristallisiert in der Wurtzit-Struktur, in der nur 4 Kationen von 4 Anionen umgeben sind und umgekehrt. Die Abbildung zeigt dementsprechend eine Struktur mit geringer Raumerfüllung, in der große Lücken sichtbar werden (das Bild ist raumfüllend gezeichnet und nicht als Schwerpunktdarstellung). Der Grund für die unerwartete Struktur ist derselbe wie bei anderen niedrigkoordinierten Strukturen auch: gerichtete Bindungen. Nicht allein die Packungsdichte ist wichtig, sondern, im Fall gerichteter Bindungen, auch eine gute Orbitalüberlappung. Bei Ammoniumfluorid sind dies Wasserstoffbrückenbindungen, die von den N-H-Funktionen ausgehen und auf jeweils eines von vier freien Elektronenpaaren des Fluorids gerichtet sind. Passend zu den 4 Wasserstoffatomen des Ammonium-Ions und den vier freien Valenzelektronenpaaren eines Fluorid-Ions wird die 4:4-Struktur des Wurtzits ausgebildet.
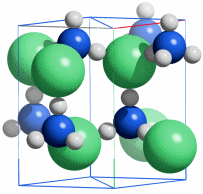
Die hexagonale Modifikation von Ammoniumfluorid (Radius des Ammonium-Ions: 150 pm, man beachte das für den CsCl-Typ passende Radienverhältnis von 0,77).
Im nun beginnenden Teil B wird ein Gang durch das Periodensystem genutzt, um die in Teil A behandelten Grundlagen anzuwenden. Der ausgearbeitete Text wird sich jetzt noch stärker auf die behandelten Beispiele beschränken, um nicht unnötig den Stoff der Lehrbücher zu wiederholen.
Es beginnt mit Wasserstoff, anschließend gehen wir von rechts nach links zuerst durch die Hauptgruppen, anschließend werden beispielhaft Aspekte der Chemie der Nebengruppenelemente behandelt, nachdem zuvor kurz Aspekte der Metallgewinnung zusammengefasst wurden.
| 1 | 2 | 13 | 14 | 15 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|
| H | He | ||||||
| Li | Be | B | C | N | O | F | Ne |
| Na | Mg | Al | Si | P | S | Cl | Ar |
| K | Ca | Ga | Ge | As | Se | Br | Kr |
| Rb | Sr | In | Sn | Sb | Te | I | Xe |
| Cs | Ba | Tl | Pb | Bi | Po | At | Rn |
| Die richtigen Lehrbuchkapitel lassen sich für die Themen des Teils B leicht finden. Sie werden daher ab jetzt nicht mehr separat bei jedem Kapitel aufgeführt. Ein knappes Lehrbuch wie der Mortimer gibt Ihnen eine Idee, welche grundlegenden Kenntnisse zu den einzelnen Stoffgruppen Sie wissen sollten. Im Skript finden Sie wieder Zusatzinformation, die in den Büchern zu kurz kommt. |
Wasserstoff hat ungewöhnliche chemische und physikalische Eigenschaften. Im Mittelpunkt der Experimente steht die Laborsynthese von Wasserstoff, der sichere Umgang mit dem spezifisch leichten und brennbaren Gas und dessen Besonderheiten, die als Folge der geringen Molekülmasse auftreten.
Um sicher mit Wasserstoff umgehen zu können – notwendige Voraussetzung für den Erfolg einer „Wasserstofftechnologie“ – sind dessen geringe Dichte sowie die stark exotherme Verbrennung zu beachten:
Versuch 3-4: Knallgas in Seifenblasen
Versuch 5-8: Dichte von H2
Versuch 5-9: Umgang mit H2
Versuch 5-16: Explosionsgrenzen
Versuch 5-20: Knallgasgebläse
Die Laborsynthese von Wasserstoff berücksichtigt die elektrochemische Spannungsreihe. Ist das verwendete Metall unedel genug, reicht das Potential des Wasserstoffs in Wasser aus, um das Metall zu oxidieren und dabei selbst zu H2 reduziert zu werden. Voraussetzung ist, dass keine Passivierung des Metalls eintritt.
Versuch 5-2: Li + Wasser
Versuch 5-3: Mg + Wasserdampf
Die bei ca. 900 °C ausgeführte technische Wasserstoffsynthese aus Methan und Wasser gemäß
CH4 + H2O → CO + 3 H2
ist ein Beispiel für eine Reaktion, welche die thermodynamischen Rahmenbedingungen klar widerspiegelt. Um dies zu analysieren, werden lediglich die Standardbildungsenthalpien ΔHf°/kJ mol−1 der gasförmigen Reaktanden benötigt: CH4 −79, H2O −242, CO −110, CO2 −394. Die Zahlenangabe für Kohlendioxid wird bei der nachfolgenden Umsetzung benötigt, der CO-Konvertierung, bei der die Einstellung des Wassergasgleichgewichts erzielt werden muss:
CO + H2O → CO2 + H2
Auch hier folgt die Reaktionsführung den Vorgaben der thermodynamischen Größen.
Bei der Handhabung von Wasserstoff ist neben seiner Brennbarkeit (siehe vor allem die Katastrophe von Lakehurst, 1937) seine extrem schnelle Diffusion zu beachten, die unter allen Gasen nur noch von Helium geteilt wird.
Versuch 5-12: H2-Diffusion in Tonzelle
Reaktionen des Wasserstoffs starten oft mit der Bindungsspaltung des recht stabilen H2-Moleküls (D = 435 kJ mol−1), so dass in der Regel eine Zündung der Reaktion nötig ist. Dies gilt nicht, wenn als Katalysator Platin oder Palladium anwesend ist. In diesem Fall startet die Reaktion oft bereits bei Raumtemperatur – offensichtlich gelingt durch diese Metalle die H-H-Bindungsspaltung unter Umgebungsbedingungen. Es liegen einige Strukturanalysen vor, welche die hohe katalytische Aktivität von Platin und Palladium deutlich machen. Die beiden Metalle kristallisieren in der kubisch dichtesten Kugelpackung. Palladium nimmt beim Kontakt mit Wasserstoff diesen bis zu einer Formel PdH0,6 auf, ohne dabei die Struktur zu verändern – von einer geringen Vergrößerung der Gitterkonstanten abgesehen. Was bei der Diskussion von Salzstrukturen als Formalismus benutzt wurde, findet hier als chemische Reaktion statt: das Füllen von Lücken in einer dichtesten Packung von Metallatomen:
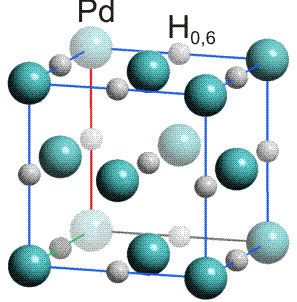
Versuch 5-13: H2 löst sich in Palladium
Wird Palladium unter drastischeren Bedingungen mit Wasserstoff umgesetzt, verändert sich die Anordnung der Metallatome:
Die gezeigte Verbindung ist mit der Formel PdH0,75 etwas wasserstoffreicher als PdH0,6. Die Palladiumatome kristallisieren nun im Wolfram-Typ. Wir machen uns klar, dass die Wasserstoffatome verzerrte Tetraederlücken besetzten und dass jede kleine Kugel im Bild 1/8 eines H-Atoms darstellt (jede Kugel trägt dann zu 1/16 zur abgebildeten Zelle bei, die gezeigten 24 Kugeln zu 24/16 = 1.5 H pro 2 Pd = PdH0.75 [2 Pd wegen 8/8 + 1 Pd pro Elementarzelle]). Die gelben Stäbe stellen keine normalen Bindungen dar, sie zeigen eher Pfade an, auf denen die Wasserstoffatome durch die Metallstruktur diffundieren können. Die Trennung der Wasserstoffmoleküle in H-Atome zusammen mit deren hoher Beweglichkeit in einer Metallstruktur vermittelt eine Vorstellung auf molekularer Größenskala, wie technische Katalyse abläuft.
PdH0,6 und PdH0,75 sind metallische Hydride. Wasserstoff ist außerdem in der Lage, mit elektropositiven Metallen wie Lithium, Natrium oder Calcium salzartige Hydride zu bilden, in denen der Wasserstoff in der Oxidationsstufe −I vorliegt. Das Hydrid-Ion in solchen ionischen Hydriden ist eine extrem starke Brønsted-Base, die beim Kontakt mit Wasser – besonders stürmisch mit Säure − sofort unter Bildung von H2 symproportioniert (aus einer niedrigen und einer höheren Oxidationsstufe in eine mittlere übergeht) – eines der wenigen Beispiele, dass eine Protolyse zugleich eine Redox-Reaktion ist:
H3O+ + H− → H2O + H2
Versuch 5-6: CaH2 + Wasser
Und zum Schluss …
Versuch 5-24: Luftballon mit Wasserstoff
Versuch 5-18: Elektrische Zündung von Knallgas
Die Halogene Fluor, Chlor, Brom und Iod bilden eine recht einheitliche Elementgruppe, innerhalb der sich die Eigenschaften von Fluor zu Iod graduell ändern: Die Reaktivität der Elemente nimmt ab, ebenso die Stabilität der Oxidationsstufe −I. Demgegenüber nimmt die Stabilität der positiven Oxidationsstufen zum Iod hin zu. „Iodsalz“ ist daher Natriumchlorid mit einer Beimengung von Natriumiodat. Untereinander bilden die Halogene die Interhalogenverbindungen – neben den Chalkogenhalogeniden typische „Lehrbuchmoleküle“ zum Einüben von Struktur-Bindungs-Beziehungen.
Die Chemie der Halogene erschließt sich in der Regel von der Oxidationsstufe −I aus. Technisch werden die Halogenide durch Elektrolyse zu den elementaren Halogenen oxidiert. Besondere Bedeutung hat dabei die Chlor-Alkali-Elektrolyse, die neben Wasserstoff Chlor und Natronlauge liefert:
2 NaCl + 2 H2O → H2 + Cl2 + 2 NaOH
Von den beiden Elektrodenreaktionen, der Oxidation von Chlorid an der Anode
2 Cl− → Cl2 + 2 e−
und der Reduktion von H+I zu H2 an der Kathode
2 H2O + 2 e− → H2 + 2 OH−
ist nur die zweite aufgrund der Lage der elektrochemischen Potentiale zu erwarten; anstelle der Chlorid-Oxidation sollte die Bildung von Sauerstoff gemäß
4 H2O → O2 + 4 e− + 4 H3O+
erfolgen, da E° für diese Reaktion kleiner ist als für die Chlor-Bildung (die Anode oxidiert zuerst die reduzierte Form mit dem kleineren Potential, vgl. +1,36 V für die Chlor-Abscheidung mit E(pH 7)/V = +1,23 − 7 · 0,059 = 0,82 für die Sauerstoffabscheidung aus einer neutralen Sole zu Beginn der Elektrolyse). Hier gelingt es durch Wahl eines Elektrodenmaterials mit höherer Überspannung für Sauerstoff, die Chlor-Bildung zu bevorzugen. Ein größeres Problem ist es, den Kontakt zwischen der an der Kathode entstehenden Natronlauge und dem anodisch gebildeten Chlor zu verhindern. Wegen der Reaktion von Chlor mit Natronlauge (siehe unten) ist eine Trennung der Elektrodenräume notwendig. Neu gebaute Anlagen verwenden hierzu ausschließlich das Membranverfahren, bei dem eine nur für Kationen durchlässige Membran den für die Elektrolyse notwendigen Ionentransport zwischen den beiden Elektrodenräumen gewährleistet.
Die Chemie der Halogene ist durch deren hohes Bestreben bestimmt, in die Oxidationsstufe −I überzugehen (vergleiche den übernächsten Versuchsblock). Umgekehrt bedarf es starker Oxidationsmittel, um aus Halogen(−I)-Verbindungen die freien Halogene herzustellen.
Versuch 14-3: Konz. Salzsäure reagiert mit Braunstein (MnO2) zu Chlor, das mit Kaliumiodid/Stärke-Papier nachgewiesen wird • MnIVO2 + 2 Cl− + 4 H3O+ → Mn2+ + Cl2 + 6 H2O
Versuch 14-7: Brom wird in der analogen Reaktion hergestellt.
Wird bei der Umsetzung von Braunstein mit NaCl-Lösung anstelle von Salzsäure wohl auch Chlor freigesetzt?
Bei der Reaktion mit Halogenen bilden sich Halogenide. Die Reaktivität der Halogene ist dabei so hoch, dass auch Edelmetalle umgesetzt werden und dass oft die höchstmögliche Oxidationsstufe erreicht wird.
Versuch 14-13: Chlor und Antimon • 2 Sb + 5 Cl2 → 2 SbCl5
Versuch 14-14: Chlor und Eisenwolle • 2 Fe + 3 Cl2 → 2 FeCl3
Versuch 14-15: Chlor und Messing, Chlor und Kupfer • Cu + Cl2 → CuCl2
Versuch 14-16: Brom und Antimon • 2 Sb + 3 Br2 → 2 SbBr3
Versuch 14-17: Brom und Aluminium • 2 Al + 3 Br2 → 2 AlBr3
Versuch 14-18: Iod und Aluminium • 2 Al + 3 I2 → 2 AlI3
Fluor nimmt als reaktivstes Element eine Sonderstellung ein, indem es mit allen übrigen Elementen des Periodensystems außer Helium, Neon und Argon Fluoride bildet, oft in der höchstmöglichen Oxidationsstufe des jeweiligen Elements.
Die beiden Versuche zeigen das im Labor und in der Technik oft angewandte Prinzip: „die schwächer flüchtige Säure vertreibt die stärker flüchtige Säure aus ihren Salzen.“ Diese blumig formulierte Regel ist ein schönes Beispiel für das Massenwirkungsgesetz. (Beachten Sie, dass nicht die Stärke der Säure der entscheidende Parameter ist.)
Versuch 15-5: HCl aus NaCl
Die Säurestärke nimmt von HF zu HI hin zu. Wie oft im Periodensystem ist der auffälligste Sprung der Eigenschaften zwischen der ersten und zweiten Achterperiode. Wir machen uns den Gang der Aciditäten durch ein simples elektrostatisches Modell klar.
Versuch 14-11: Iod in Wasser, KI-Lösung, Benzin, und Ethanol
Das aus völlig unpolaren Molekülen aufgebaute und daher hydrophobe Iod ist erwartungsgemäß in unpolaren Lösungsmitteln löslich, in Wasser dagegen praktisch unlöslich. Die Löslicheit in wässriger Kaliumiodid-Lösung ist daher völlig unerwartet. Den Schlüssel zum Verständnis dieses Phänomens gibt uns eine Strukturanalyse an dunkelbraunen Kristallen, die aus solchen Lösungen gewonnen werden konnten. Deren Formel ist KI3 · H2O; neben Kaliumionen und Wassermolekülen liegen Triiodid(1−)-Ionen, I3−, vor. Die Löslichkeit erklärt sich damit in der Umwandlung des hydrophoben I2-Moleküls in ein wasserlösliches Salz. Die I3−-Anionen sind linear aufgebaut:
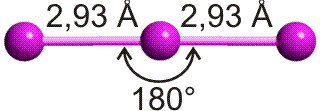
Die beiden gleich langen I-I-Atomabstände sind länger als im I2 (272 pm in festem Iod). Aufbau und Struktur des Anions sind sehr überraschend. Bei der Diskussion der chemischen Bindung traten bislang keine Spezies auf, bei denen ein Atom in seiner Valenzschale das Elektronenoktett überschreitet. Genau dies aber müsste angenommen werden, wenn mit den üblichen 2-Elektronen-2-Zentren-Bindungen formuliert würde:
Bei konventionellem Gebrauch der Valenzbindungsmethode würde man die Elektronenkonfiguration des mittleren Iodatoms mit sp3d angeben; es würde also ein d-Orbital hinzugenommen, um die zehn Elektronen am mittleren Iod unterzubringen. Nicht nur die Strukturanalyse, die eine geringere I-I-Bindungsordnung nahelegt, sondern auch die theoretische Behandlung der Bindung zeigt, dass die Beteiligung von d-Orbitalen nur untergeordnete Bedeutung hat (in einer Lewis-Formulierung: … die nur mesomere Grenzstrukturen mit geringem Gewicht hinzufügt). Wir berücksichtigen dies in der Vorlesung, indem wir in der Nichtmetallchemie stets die Oktettregel beachten.
Versuche, die Elektronenanordnung durch I3− in diesem Sinne realistischer zu deuten, sollten daher berücksichtigen: (1) das Oktett nicht zu überschreiten und (2) die aus den Bindungslängen hervorgehende Verminderung der Bindungsordnung zu erklären. Hier ein Versuch mit Lewis-Formeln, bei denen die kovalente Bindungsordnung ½ ist und das Oktett durch die Beteiligung eines ionischen Bindungsanteils erhalten bleibt:
Die beiden Grenzstrukturen beschreiben eine 4-Elektronen-3-Zentren-Bindung:
Eine solche Bindung wurde bereits behandelt, und zwar bei der starken Wasserstoffbrückenbindung im HF2−-Ion:
Die kovalente Bindungsordnung ergibt sich aus den Lewis-Formeln zu ½. Zu demselben Ergebnis führt eine Betrachtung der Molekülorbitale. Ein MO-Schema wird erhalten, indem jeweils 1 Atomorbital bei jedem Bindungspartner für das MO-Schema herangezogen wird (2p bei Fluor, 1s bei Wasserstoff). Kombination der drei Atomorbitale führt zu einem bindenden, einem nichtbindenden und einem antibindenden Molekülorbital; schematisch:
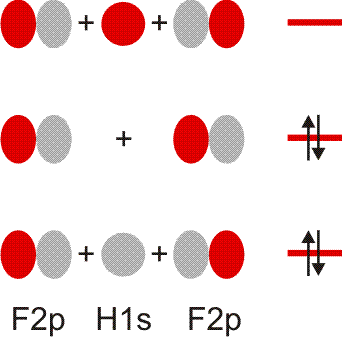
Das bindende und das nichtbindende MO sind mit jeweils zwei Elektronen besetzt. Die drei Zentren sind also durch ein Elektronenpaar verbunden, für den einzelnen F-H-Kontakt ergibt sich damit die Bindungsordnung ½. Die quantitative MO-Rechnung ergibt für die drei Orbitale:
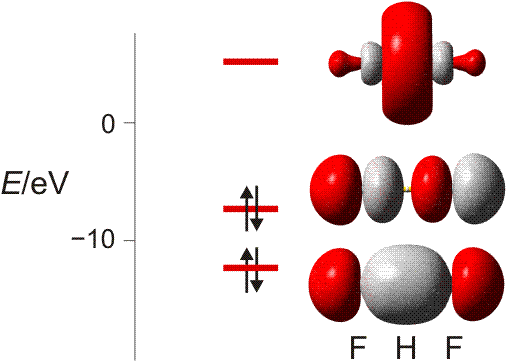
Die 4e-3z-Bindung im HF2−-Ion. Technische Anmerkung: Energien und Orbitale mit mp2/aug-cc-pvqz außer Gestalt des antibindenden Orbitals, diese mit hf/6-31g.
Im I3−-Ion ist auch das mittlere Orbital vom p-Typ. Schematisch:
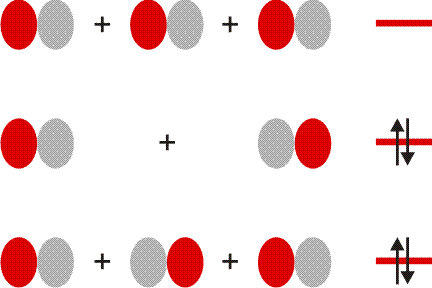
Die Bindungsordnung des einzelnen I-I-Kontakts ist wieder ½ – im Einklang mit der Lewis-Formulierung und den experimentell bestimmten Atomabständen.
Beim der Umsetzung gleicher molarer Mengen zweier Halogene bilden sich alle sechs denkbaren zweiatomigen Interhalogen-Verbindungen. So entsteht Chlorfluorid, ClF, als farbloses Gas gemäß
Cl2 + F2 → 2 ClF ΔrH° = −113 kJ mol−1
bei der exothermen Reaktion eines Cl2/F2-Gemischs bei Temperaturen unterhalb ca. 300 °C unter Kupferkatalyse.
Werden anstelle des 1:1-Gemischs 3 Teile Fluor und 1 Teil Chlor eingesetzt, so entsteht Chlortrifluorid, ebenfalls als farbloses Gas:
Cl2 + 3 F2 → 2 ClF3 ΔrH° = −330 kJ mol−1
Es können weitere Halogenfluoride hergestellt werden: neben BrF3 und IF3 die Halogen(V)-fluoride ClF5, BrF5 und IF5, sowie IF7, ferner ICl3.
Wie lässt sich die chemische Bindung in diesen Molekülen deuten? Bei ClF3 müsste die Valenzschale des Chloratoms Platz für 7 + 3 = 10 Elektronen aufbringen, wenn ausschließlich 2e-2z-Bindungen formuliert würden – wieder müssten d-Orbitale im Sinne einer sp3d-Hybridisierung beteiligt werden. Wie beim I3− zeigt die theoretische Behandlung jedoch eine nur geringe d-Orbital-Beteiligung an, so dass die Näherung „keine Beteiligung“ realistischer ist als „volle Beteiligung“. Solange wie zum Beispiel bei IF7 nicht die höchste Oxidationsstufe erreicht ist, wennn also noch mindestens 1 freies Elektronenpaar in der Valenzschale des Zentralatoms vorliegt, zeigen dieselben Rechnungen, dass das dann mit 2 Elektronen gefüllte s-Orbital als freies Elektronenpaar vorliegt und nur wenig zu den Bindungen beiträgt. Unter diesen beiden (etwas, aber nicht dramatisch) vereinfachenden Voraussetzungen – Bindungen nur mit p-Orbitalen des Zentralatoms, keine s-, keine d-Orbital-Beteiligung – kann die Struktur einer Interhalogenverbindung vorausgesagt werden.
Als Beispiel soll ClF3 eingehender behandelt werden. Die experimentelle Strukturbestimmung führt zu einer T-förmigen Gestalt des Moleküls:
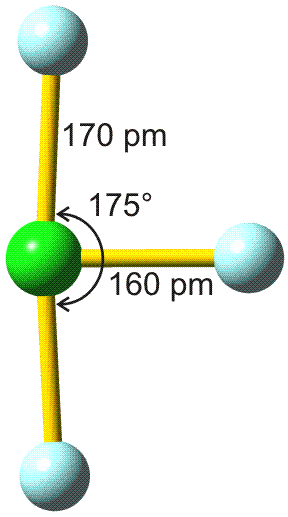
In der annähernd linearen F-Cl-F-Einheit sind die Fluoratome etwas zum dritten F-Atom hingeneigt. Die Cl-F-Abstände sind unterschiedlich: länger im annähernd linearen F-Cl-F-Fragment, kürzer bei der dritten Cl-F-Bindung.
Diese Struktur kann mit Hilfe des folgenden „Rezeptes“ vorausgesagt werden:
• (noch 'mal zur Sicherheit:) liegt das Zentralelement in der höchsten Oxidationsstufe vor? Wenn nein, so wie hier, weiter:
• Bilde 2e-2z-Bindungen. Notiere hierzu die Lewis-Formel des Zentralatoms und binde so viele Substituenten wie möglich bis das Oktett erreicht ist. Stelle die Orbitale der freien Elektronenpaare dar (das doppelt besetzte s-Orbital ist nur deswegen gezeichnet damit nichts fehlt, man kann es bis zur Lewis-Formel auch weglassen):
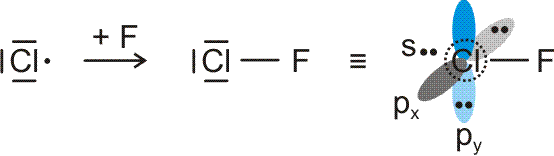
• bilde 4e-3z-Bindungen mit den restlichen Substituenten:
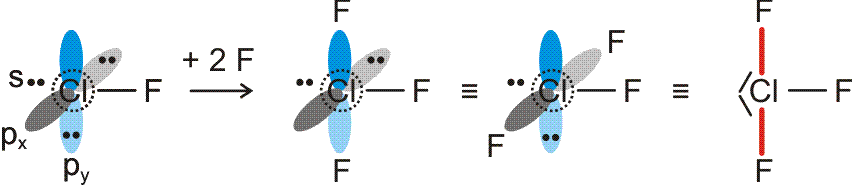
In eine Lewis-Formel übertragen, ergibt sich:
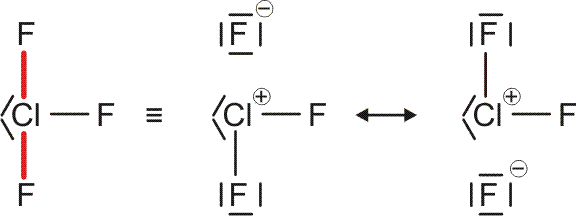
Die Lewisformeln verdeutlichen die Gültigkeit der Oktettregel sowie die Ungleichheit der Cl-F-Bindungen, die das Ergebnis der Strukturanalyse war: in der 4e-3z-Bindung ist die kovalente Bindungsordnung ½.
Die gezeigten mesomeren Grenzstrukturen werden nur dann großes Gewicht haben, wenn die Atome an den Enden der annähernd linearen X-Cl-X-Einheit hinreichend elektronegativ sind. Die Bedingung wird nicht mehr erfüllt, wenn die F-Atome zum Beispiel gegen H-Atome ausgetauscht werden. Es gibt daher HCl, nicht aber die zu ClF3, ClF5 und ClF7 analogen Moleküle H3Cl, H5Cl und H7Cl.
Sie können das beschriebene Verfahren nicht nur auf Interhalogene anwenden (siehe die nächsten Kapitel) und auch nicht nur auf neutrale Verbindungen, sondern auch auf Ionen.
Und jetzt zum unangenehmen Teil. Es gibt natürlich den Bedarf, den Aufbau eines Moleküls unkompliziert hinzuzeichnen; das heißt, zum Beipiel, ohne mesomere Grenzformeln. Es ist daher üblich, den Aufbau von ClF3 durch die Formel wiederzugeben:
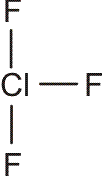
Dadurch wird natürlich unsere ganze Diskussion auf den Kopf gestellt. Wenn Sie eine solche Formel sehen, haben Sie zwei Möglichkeiten, diese aufzufassen: (1) sie denken sich: hier ist eine mesomere Grenzstruktur mit d-Orbital-Beteiligung aufgestellt; ich weiß, dass diese nur einen kleinen Anteil hat; sie wurde gewählt, weil sie am einfachsten hinzuzeichnen ist. (2) Sie begreifen diese Formel als topologische Formel, die nur den Aufbau einer Verbindung wiedergibt sowie eine Aussage darüber macht, welche Atome mit welchen irgendwie miteinander verbunden sind. Kürzer: ein Strich ist nicht immer eine 2e-2z-Bindung.
Alle Halogene außer Fluor bilden Verbindungen, in denen sie in positiven Oxidationsstufen auftreten. Ein wichtiges Beispiel ist die Umsetzung von Chlor mit Natronlauge, die bei der Chlor-Alkali-Elektrolyse gerade vermieden wird. In der Hitze entsteht dabei in einer Disproportionierung Chlorat(V) gemäß:
3 Cl2 + 6 OH− → ClO3− + 5 Cl− + 3 H2O
In der Kälte hingegen führt die Disproportionierung nur bis zur Stufe von Chlorat(I), „Hypochlorit“:
Cl2 + 2 OH− → ClO− + Cl− + H2O
Versuch 19-1: Reaktion von Chlor mit Natronlauge
Bei niedrigerem pH-Wert liegt eine typische Gleichgewichtsreaktion vor:
Cl2 + 3 H2O ⇄ ClO− + Cl− + 2 H3O+
Versuch ?: Bleichen von Lackmuspapier durch Hypochlorit beim CO2-Überleiten
Diese Umsetzung ist für die pH-Abhängigkeit der Desinfektionswirkung des Chlors verantwortlich. Ein saures (kohlendioxidreiches) Wasser benötigt weniger Chlor bis zur Entkeimung als ein alkalisches Wasser, da ClO− kaum desinfiziert. (Vielleicht kennen Sie die handelsüblichen Desinfektionslösungen für Swimmingpools: hier wird Natriumhypochlorit mit einer genau angegebenen Menge Salzsäure gemischt.)
Beim Entwerfen der Lewis-Formel von Chlorat(V) begegnet uns ein bereits diskutiertes Problem: die Oktettaufweitung in vielen Büchern; auch hier formulieren wir im Einklang mit der Oktettregel gemäß
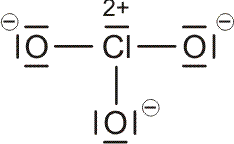
Halogenate haben mit den Halogenen gemeinsam, dass sie in oft heftiger Reaktion als Oxidationsmittel wirken. Lehramtsstudierende und künftige Apotheker sollten die folgenden Reaktionen kennen. Sie wissen dann, welche Chemikalien sie nicht aus der Hand geben sollten:
Versuch 19-11: Kaliumchlorat(V) und Aluminium
Versuch 19-12a: Kaliumchlorat(V) und Schwefel
Versuch 19-12b: Kaliumchlorat(V) und roter Phosphor
Die oxidierende Wirkung der Halogenate(V) nimmt zum Iod hin so stark ab, dass Alkaliiodate(V) und nicht Alkaliiodide bei Lagerung an der Luft stabil sind („Iodsalz“ enthält Kaliumiodat). Es gelingt aber die Symproportionierung zu Iod:
Versuch 19-14: Kaliumiodat(V) und Kaliumiodid
Eine technisch wichtige, aber doch brisante Verbindung ist Chlor(IV)-oxid (Chlordioxid). ClO2 gehört neben NO und NO2 zu den wenigen Radikalen, die als Substanz isolierbar sind:
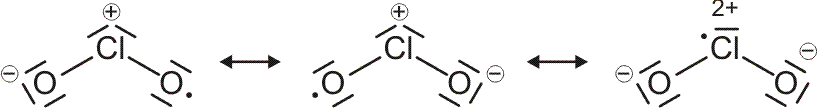
Die mesomeren Grenzformeln haben annähernd das gleiche Gewicht, wie sich durch Rechnung zeigen lässt. Das ungepaarte Elektron besetzt das höchste besetzte Molekülorbital (HOMO), ein Cl-O-antibindendes Orbital vom π-Typ, mit annähernd gleichen Beiträgen der zugrunde liegenden Atomorbitale; im Bild liegt das Chloratom vorne, die beiden Sauerstoffatome weisen nach hinten:
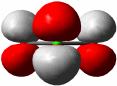
Die Herstellung von ClO2 umfasst einige der besprochenen Reaktionen. Im ersten Schritt wird NaCl-Lösung in einer ca. 80 °C heißen Sole elektrolysiert:
NaCl + 3 H2O → NaClO3 + 3 H2
Anschließend wird mit HCl umgesetzt:
2 NaClO3 + 4 HCl → 2 ClO2 + Cl2 + 2 NaCl + 2 H2O
Die Umsetzung wird möglich, da die bei der Protolyse von Chlorat(V) gebildete Chlor(V)-Säure unbeständig ist und disproportioniert. Chlordioxid wird auf diesem Weg in großen Mengen als Bleichmittel hergestellt (näheres im „Kapitel 16. Hauptgruppe“).
Versuch 19-17: Herstellung und Zersetzung von Chlordioxid
Die Edelgase Helium, Neon, Argon, Krypton und Xenon haben Bedeutung in Technik und Labor, da sie keine oder geringe Neigung haben, Verbindungen mit irgendeinem Raktionspartner einzugehen. So ist das recht häufige Element Argon, das zu ca. 1 % in der Luft enthalten ist, ein geläufiges „Schutzgas“, das reaktive Stoffe vor dem Kontakt mit Luft schützen soll („Argonschweißen“). Eine etwas umfangreichere Chemie hat sich lediglich mit Xenon entwickelt.
XeF2, XeF4, XeF6 sowie das sehr unbeständige KrF2 entstehen bei der direkten Umsetzung der Elemente. Im Gegensatz zu Kryptondifluorid ist Xenondifluorid eine stabile Verbindung, die als farbloses kristallines Pulver kommerziell erhältlich ist.
Die im letzten Kapitel eingeführte 4e-3z-Bindung bestimmt die Strukturen und die Bindungsverhältnisse der Edelgasfluoride. Da bereits die Edelgasatome über ein Valenzelektronenoktett verfügen, gibt es keine 2e-2z-Bindungen in den Verbindungen der Edelgase. Das oben beschriebene Verfahren zur Vorhersage der Struktur beschränkt sich daher darauf, die entsprechende Zahl von Fluorpaaren an jeweils ein p-Orbital anzulagern. So ergibt die Vorhersage für Xenon(II)-fluorid eine lineare Struktur:
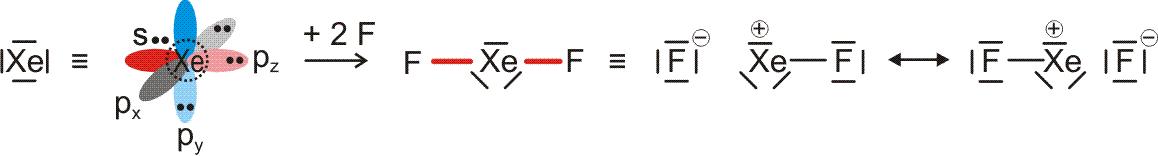
Für Xenon(IV)-fluorid wird auf demselben Weg quadratisch-planarer Aufbau vorhergesagt:
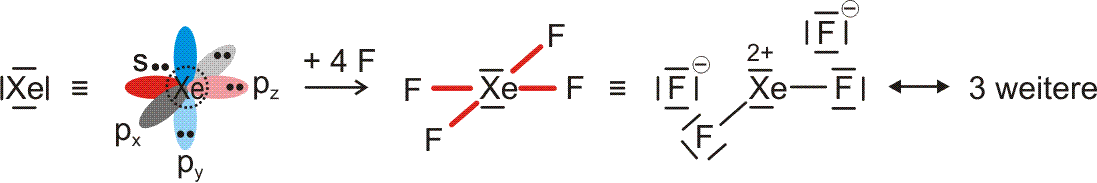
Das Experiment bestätigt beide Vorhersagen. Man beachte, dass sich wegen derselben Valenzelektronenanordnung dieselben Strukturen für ClF4− und für XeF4 ergeben.
Neben Oxidfluoriden sind von Xenon zwei Oxide bekannt, XeO3 und XeO4, die beide positive Standardbildungsenthalpien aufweisen und explosionsartig zerfallen können. Die Formeln lassen sich im Einklang mit der Oktettregel aufstellen und entsprechen völlig dem isoelektronischen Iodat(V), IO3−, und Iodat(VII), IO4−, und auch der Aufbau der entsprechenden Spezies ist gleich: trigonale Pyramide und Tetraeder.
Von besonderem Interesse ist der Bezug zur wässrigen Chemie: beide Oxide sind Anhydride von Säuren. Xenon(VI)-Säure, H2XeO4, bildet sich beim Lösen von XeO3 in Wasser, XeO4 lässt sich aus Xenat(VIII)-Salzen, die das [XeO6]4−-Ion enthalten, herstellen. Um ein solches „Perxenat“ so formulieren, sind nun wieder 4e-3z-Bindungen nötig, um im Einklang mit der Oktettregel zu bleiben.
Die Chalkogene Sauerstoff, Schwefel, Selen und Tellur zeigen eine viel deutlichere Abstufung ihrer Eigenschaften als die Halogene. Ein weiterer Unterschied ist das Vorkommen der freien Elemente Sauerstoff und Schwefel – Sauerstoff in der Luft und Schwefel in mineralischer Form. Besonders interessant: das Auftreten der beiden freien Elemente auf der Erde ist durch Organismen verursacht.
Elementarer Sauerstoff, im Grundzustand als Triplett-Sauerstoff, in zwei angeregten Zuständen als reaktiver Singulett-Sauerstoff, wurde in der Vorlesung bereits behandelt. Auch die stabilste Oxidationsstufe des Sauerstoffs, −II, war bereits in einigen Verbindungen vertreten. Nachzutragen bleibt aber eine weitere instabile und äußerst reaktive Form des elementaren Sauerstoffs, Ozon. Wegen der großen praktischen Bedeutung sollen ferner die Peroxide behandelt werden. Die Stammverbindung ist das Wasserstoffperoxid, H2O2, die Oxidationsstufe des Sauerstoffs ist hier −I.
Ozon, O3, ist eine reaktive allotrope Form des Elementes Sauerstoff. Ozon entsteht als unstabile Modifikation des Sauerstoffs bei der Zufuhr einer hinreichend hohen Energie (ΔfH° = +143 kJ mol−1). In der Stratosphäre wird die erforderliche Energie durch UV-Strahlung zur Verfügung gestellt (hier dient Ozon aufgrund seiner hohen Absorption für harte UV-Strahlung als Schutz vor energiereicher Strahlung), während die Ozonbildung in Bodennähe, also bei geringerer Strahlungsenergie, NO-katalysiert verläuft. In beiden Fällen und auch beim Betrieb eines „Ozonisators“ werden O2-Moleküle durch Strahlungsenergie oder elektrische Energie gespalten. Die entstandenen O-Atome reagieren dann gemäß
O + O2 → O3
Die Elektronenanordnung im Ozon-Molekül wird durch mesomere Grenzformeln wiedergegeben (wir machen uns die Bedeutung der rechten Formel durch einen Vergleich mit Schwefeldioxid klar):
Versuch 10-1: Ozon aus Ozongenerator, Nachweis mit KI-Lösung, Phenolphthalein und Stärkelösung.
• Ozon oxidiert Iodid gemäß O3 + 2 I− + H2O → I2 + 2 OH− + O2
Versuch 10-2: Zersetzung von Ozon durch Erhitzen
• 2 O3 → 3 O2
Versuch 10-3: Katalytische Zersetzung von Ozon
Versuch 10-5: Ozon und Silber
• bei der Reaktion von Ozon mit Silber entstehen schwarze Silberoxide
2 Ag + [O] → Ag2O und Ag + [O] → AgO. AgO ist nicht Silber(II)-, sondern Silber(I,III)-oxid. [O] bedeutet, dass der Sauerstoff in nicht näher bekannter Weise aus Ozon stammt. Obwohl der Weg O3 → [O] + O2 naheliegt, gibt es doch Hinweise, dass der entstehende Sauerstoff, möglicherweise Singulett-Sauerstoff, ebenfalls reagiert, so dass gilt: O3 → 3 [O].
Versuch 10-6: Bleisulfid und Ozon
• PbS + 4 [O] → PbSO4
Versuch 10-7: Oxidation von Mangan(II) durch Ozon zu Braunstein
• MnSO4 + [O] + 3 H2O → MnO2 + SO42− + 2 H3O+
Versuch 10-8: Ozon zerstört Indigocarmin
Versuch 10-10: Reaktion organischer Lösungsmittel mit Ozon
Die beiden letzten Reaktionen verweisen auf eine wichtige Anwendung, nämlich die Verwendung von Ozon als Desinfektionsmittel. Zusammengenommen belegen die Versuche, dass Ozon eines der stärksten bekannten Oxidationsmittel ist; das Standardpotential der Halbzelle O3 + 2 H3O+ + 2 e− ⇄ O2 + 3 H2O beträgt 2,07 V in saurer Lösung (berechnen Sie das Potential bei pH 7). Damit übereinstimmend ist Ozon ein die Atemwege reizendes Gas.
In der Oxidationsstufe −I liegen mit den Peroxiden reaktive Sauerstoffverbindungen vor, die in großem Umfang praktische Anwendung finden, aber auch von unmittelbarer Bedeutung für Organismen sind.
Ein Zugang zu Peroxiden besteht in der überraschenden Reaktion von Natrium-Metall beim Verbrennen an der Luft. Anstelle des erwarteten Produkts Natriumoxid bildet sich Natriumperoxid, Na2O2. Neben Natrium-Ionen liegen in diesem gelben Salz O22−-Ionen vor. Peroxid-Ionen reagieren in charakteristischer Weise mit einigen Übergangsmetallionen zu Peroxido-Komplexen; Beispiele werden mit Titan(IV) und mit Chrom(VI) gezeigt. Die Reaktion mit Titan(IV) ist ein geläufiger Nachweis für Peroxid.
Versuch 18-14: Herstellung von Natriumperoxid und Nachweis des Peroxids mit Titan(IV)
• 2 Na + O2 → Na2O2
Versuch 18-13: Blaues CrO5
• CrO42− + 2 H2O2 + 2 H3O+ → CrO(O2)2 + 5 H2O
Versuch 18-15: Oxidation von Aluminium mit Natriumperoxid
Mit Chlor, Chlordioxid, Ozon und Wasserstoffperoxid wurden starke Oxidationsmittel beschrieben, die aufgrund ihrer aggressiv oxidierenden Wirkung als Desinfektionsmittel und/oder Bleichmittel verwendet werden.
Mengenmäßig besonders bedeutend ist der Einsatz von Bleichmitteln in der Zellstoffherstellung. Zellstoff ist der Celluloseanteil des Holzes. Ein letzter Schritt zu dessen Gewinnung in der Papierherstellung ist die Bleiche, bei der nicht abgetrennte Reste des braun färbenden Holzbestandteils Lignin zu farblosen Reaktionsprodukten abgebaut werden. Die Bleiche mit Chlor gilt als wenig umweltfreundlich, zum Beipiel verbleiben hierbei im Endprodukt chlorierte organische Substanzen, die je nach Verwendungszweck als bedenklich erscheinen (zum Beispiel bei Filterpapieren für Kaffee oder Tee, die heiß extrahiert werden und das Produkt anschließend verzehrt wird).
Chlordioxid hat nicht nur den Vorteil, in geringerer Menge wirksam zu sein, die Bildung chlorierter organischer Verbindungen ist hier weitgehend zurückgedrängt. Die entsprechenden Produkte werden unter der Bezeichnung elementarchlorfrei gebleicht vermarktet.
Versuch 18-3: Nachweis von Peroxid in Waschmittel
Versuch 18-8: Eisenkatalysiertes Bleichen von Indigo
Die Peroxidbleiche ist das Standardverfahren bei der Textilwäsche. Da Peroxide recht hohe Temperaturen benötigen um ihre Bleichwirkung zu entfalten (70–80 °C), das „Kochen“ der Wäsche aber wegen des hohen Energieverbrauchs aus der Mode gekommen ist, enthalten moderne Waschmittel Bleichverstärker. Dies sind Übergangsmetallverbindungen, die das Peroxid aktivieren. Ein Beipiel, dass auch in der organischen Synthese gezielt eingesetzt wird, ist die Verwendung von Eisen(II) in der „Fenton-Reaktion“:
H2O2 + Fe2+ → [FeIII(OH)]2+ + HO•
Wasserstoffperoxid ist durch diese Reaktion in das viel reaktionsfähigere Hydroxyl-Radikal umgewandelt worden.
Neben den Peroxiden kann die unvollständige Reduktion von Sauerstoff zu Hyperoxid führen (in der Biochemie meist als Superoxid bezeichnet). In reiner Form entsteht Hyperoxid bei der Verbrennung von Kalium und seinen schweren Homologen Rubidium und Cäsium an der Luft:
K + O2 → KO2
Das außerordentlich reaktive Hyperoxid-Ion ist ein Radikal-Anion:
Die lehrbuchübliche Lewis-Formulierung zeigt korrekt an, dass im Anion ein ungepaarter Spin vorliegt, die Bindungsordnung wird nicht richtig wiedergegeben. Die Frage lässt sich klären, wenn auf das MO-Schema des Sauerstoffs zurückgegriffen wird. Die Rechnung zeigt für das Radikalanion als Folge der Aufnahme eines Elektrons etwas destabilisierte Orbitale an. Der wesentliche Unterschied betrifft aber die Besetzung der Valenzorbitale: 13 statt 12 Elektronen führen zur Besetzung der antibindenden 2π-Orbitale durch 3 Elektronen und damit zu einer Bindungsordnung von
BO = ½ (8 − 5) = 1,5
Die O-O-Bindungsordnung liegt somit zwischen den Werten von zwei in O2 und eins in H2O2. Wird für die Lewis-Formel Paulings Dreielektronen-Bindung verwendet, wird auch die Bindungsordnung korrekt angezeigt:
Aerob lebende Organismen gewinnen ihre Energie aus der Atmungskette. Das letzte Enzym der Atmungskette, Cytochrom-c-Oxidase, reduziert ein O2-Molekül ohne Zwischenschritte durch 4 Elektronen zu Wasser:
O2 + 4 e− + 4 H+ → 2 H2O
Wird Sauerstoff auf dem Weg zur Cytochrom-c-Oxidase durch einen der zahlreichen 1-Elektronen- (Cytochrome, Ferredoxine, NADH) oder 2-Elektronenüberträger (FADH2) des Organismus unvollständig reduziert, so entstehen reaktive Sauerstoffspezies. Es ist eine Schätzung publiziert, nach der ca. 5 % der Elektronen der Atmungskette fehlgeleitet werden und zu Hyperoxid und Peroxid führen („oxidativer Stress“). Damit übereinstimmend können solche Spezies auch bei normalen Verbrennungsvorgängen nachgewiesen werden.
Versuch 18-1: Bildung von H2O2 in einer Wasserstoffflamme
Aerob lebende Organismen verfügen über wirksame Mechanismen, reaktive Sauerstoffspezies zu entfernen. Grundlage ist die Unstabilität dieser Spezies; sowohl H2O2 als auch Hyperoxid zerfallen exotherm:
2 H2O2 → 2 H2O + O2 ΔrH° = −196 kJ mol−1
Hyperoxid ist so reaktiv, dass es in Wasser augenblicklich Sauerstoff und H2O2 bildet:
2 O2•− + 2 H3O+ → O2 + H2O2 + 2 H2O
Trotz dieser schnellen Umsetzung setzen Organismen Superoxiddismutasen ein. Dies ist nötig, da die Geschwindigkeit des Hyperoxidzerfalls dem Quadrat der Hyperoxidkonzentration proportional ist – es ist eine Reaktion zweiter Ordnung. (Denken Sie darüber nach, was dies für die sehr kleinen physiologischen Konzentrationen bedeutet.) H2O2, das unmittelbar oder durch Superoxiddismutase-Aktivität gebildet wurde, wird durch verschiedene Katalasen gemäß der oben angegebenen Gleichung zu Sauerstoff und Wasser umgesetzt.
Versuch 3-15: Katalytische H2O2-Zersetzung (Rinderblut-Variante)
Zeigten Fluor und Chlor noch eine weitgehende Ähnlichkeit, so zeigt sich beim Paar Sauerstoff/Schwefel besonders drastisch, dass der Unterschied zwischen einem Element und seinem schweren Homologen besonders groß zwischen der ersten und zweiten Achterperiode ausfällt. Beim weiteren Gang in der Gruppe nach unten zeigen sich nur noch graduelle Unterschiede auf, vor allem eine Zunahme des metallischen Charakters innerhalb einer Gruppe.
Versuch 22-2a: Kristalline Schwefelmodifikationen aus der Schmelze
Die thermodynamisch stabile Form des Schwefels ist der im orthorhombischen Kristallsystem kristallisierende α-Schwefel („rhombischer Schwefel“), der bei 96 °C in den unstabilen β-Schwefel („monokliner Schwefel“) überzugehen beginnt. Beide Schwefelmodifikationen bestehen aus gewellten, kronenförmigen S8-Molekülen, im oberen Bild in der Aufsicht:
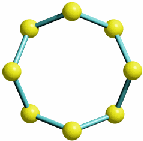
… und von der Seite:
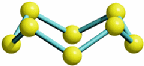
Abstände und Winkel in den S8-Molekülen unterscheiden sich kaum (Mittelwerte für α-Schwefel: S-S 205 pm, S-S-S 108°).
In der Kristallstruktur von α-Schwefel sind rechtwinklig zueinander angeordnete Schwefelringe zu erkennen:
In der Struktur der Kristallnadeln von monoklinem Schwefel fehlt dieses charakteristische Merkmal:
Dieser deutliche strukturelle Unterschied ist zu bedenken, wenn die Polymorphie von S8 näher betrachtet wird. Einige weitere Daten:
| α-S8 | β-S8 | |
|---|---|---|
| Schmelzpunkt/°C | 113 | 120 |
| ρ/g cm−3 | 2,06 | 2,00 |
| ΔfH°/kJ mol−1 | 0 | 3,2 |
| Farbe bei RT | gelb | fast fbl. |
Sowohl die Schmelzpunkte als auch der Wert für die Dichte des monoklinen Schwefels variieren je nach Publikation um einige Grad bzw. um einige Hundertstel g cm−3. Die Ursache für die Unsicherheit bei der unstabilen Form ist eine Beimischung von Bruchstücken von S8-Ringen, da die Ringe bei der recht hohen Untersuchungstemperatur zum Teil bereits gespalten sind. Der Schmelzpunkt der stabilen Modifikation ist dagegen unsicher, da deren Stabilitätsbereich bei ca. 96 °C endet. Oberhalb dieser Temperatur beginnt sich der rhombische Schwefel in die monokline Phase umzuwandeln. Diese Umwandlung verläuft im Bereich von Minuten und nicht schlagartig. Dasselbe gilt für den umgekehrten Weg: monokliner Schwefel wandelt sich unterhalb von 96 °C in rhombischen Schwefel um, wobei dieser Vorgang bei Raumtemperatur Stunden bis Wochen in Anspruch nimmt. Die Ursache für das Phasenumwandlungsverhalten geht aus den Strukturbildern hervor: die einzelnen Moleküle müssen – im Feststoff! – recht große Wege zurücklegen, um von der einen in die andere Modifikation überzuwechseln. Dies erklärt nicht nur die Dauer der Umwandlung, sondern auch die Beobachtung, dass Kristalle der einen Form bei der Transformation in die andere Form zerstört werden: nadelförmige, farblos-klare Kristalle des monoklinen Schwefels behalten bei der Phasenumwandlung ihre äußere Form, werden jedoch trübe, da sie in eine große Zahl mikroskopisch kleiner Kristalldomänen des rhombischen Schwefels zerfallen.
Es wird (erneut) deutlich, dass nicht nur die Stabilität der einzelnen Formen das Verhalten einer Substanz bestimmt, sondern auch die Aktivierungsschwellen für die jeweiligen Umwandlungen. Die Untersuchung der Stabilität bildet dabei den thermodynamischen Rahmen. Die Thermodynamik der Phasenumwandlungen von S8 lässt sich in einem Druck-Temperatur-Diagramm darstellen. Die folgende Abbildung zeigt den Ausschnitt, der den Randbereich zur Gasphase beschreibt. Der auf der Ordinate abgetragene Druck ist damit der Dampfdruck. Die folgende Betrachtung gilt daher für Schwefel unter seinem eigenen Dampfdruck (eine Schwefelprobe, die unter Vakuum in ein Probegefäß eingeschmolzen wurde):
Es sind einige Punkte hervorgehoben. T1 ist die Umwandlungstemperatur der beiden festen Phasen, T2 ist der Schmelzpunkt der α-Phase, T3 der Schmelzpunkt der β-Phase. T2 sollte unerreichbar sein, wenn die Phasenübergänge nicht durch Aktivierungsbarrieren behindert wären. So gelingt es durch schnelles Erhitzen, α-Schwefel zu schmelzen, bevor die fest-fest-Phasenumwandlung in größerem Umfang abgelaufen ist („Sofortschmelzpunkt“ mit Hilfe einer „Kofler-Bank“). Umgekehrt kann aus einer Schwefelschmelze bei sehr vorsichtigem Abkühlen unter T2 gegebenfalls rhombischer Schwefel auskristalliert werden. Im Normalfall aber wird aufgrund der im Diagramm beschriebenen Rahmenbedingungen zuerst die unstabile Modifikation auskristallisieren, die sich dann anschließend in die stabile Form umwandelt (Ostwaldsche Stufenregel).
Die α/β-Umwandlung verläuft bei Schwefel in beide Richtungen, sie verläuft als Folge des Phasendiagramms enantiotrop. Vor allem für die Herstellung fester Pharmaka erheblich wichtiger ist der Fall der monotropen Phasenumwandlung. Dieser liegt das folgende p-T-Diagramm zugrunde:
Eine unstabile Phase (fest-II) ist dabei so unstabil, dass es für sie keinen Beständigkeitsbereich gibt. Die stabile Phase (fest-I) wird sich also nie in die unstabile Phase umwandeln. Die unstabile Phase kann jedoch unter Umständen durch Abkühlen der Schmelze erreicht werden. Ihre Lebendauer ist unbestimmt, irgendwann wird sie sich in die stabile Phase umwandeln. „Monotrop“ sagt genau dies aus, dass nämlich nur die Richtung unstabil zu stabil möglich ist. Da pharmakologisch wichtige Parameter – über die Löslichkeit und/oder über die damit gekoppelte Lösungsgeschwindigkeit zum Beispiel die Bioverfügbarkeit – von der jeweiligen Modifikation wirklich oder zumindest juristisch abhängen können, hat die Kontrolle der Polymorphie bei der Entwicklung fester Arzneistoffe einen besonderen Stellenwert (Schlagwörter: Polymorphie-Screening, crystal engeneering).
Versuch 22-2b: Kristalline Schwefelmodifikationen aus Lösungen in Toluol und Schwefelkohlenstoff (CS2)
Molekular aufgebaute Stoffe werden in der Regel nicht aus der Schmelze, sondern aus Lösung kristallisiert. Hierbei gelten ähnliche Regeln wie bei der Kristallzucht aus der Schmelze. Es gilt, sowohl die Kristallkeimbildung als auch das Keimwachstum zu kontrollieren. Die für die Keimbildung benötigte Aktivierungsenergie wird auch Keimbildungsarbeit genannt. Sie ist aufzubringen, um hinreichend viele Moleküle gegen den Einfluss der Entropie zu einem Kristallkeim zusammenzulagern. Die Größe dieser Aktivierungsbarriere ist für den Verlauf der Kristallisation bedeutend. Hier nur eine Faustregel, die hier ohne nähere Begründung benutzt wird: je größer die Energiedifferenz zwischen Kristall und Lösung, desto höher die Aktivierungsenergie. Diese Regel sagt voraus, dass (1) unstabile Modifikationen auch aus Lösung bevorzugt kristallisieren (die Ostwaldsche Stufenregel gilt auch hier), und (2) dass die Chance, eine unstabile Modifikation zu kristallisieren umso größer ist, je unstabiler die Lösung gegenüber dem Feststoff ist. Da die Stabilität der Lösung oft mit der Löslichkeit eines Stoffes im Lösungsmittel korreliert, gilt weiter, dass die Chance, eine unstabile Form zu kristallisieren, aus dem schlechteren Lösungsmittel größer ist. So grob solche Faustregeln auch sind, so geben sie doch eine Vorstellung davon, warum Schwefel aus dem schlechten Lösungsmittel Toluol (ca. 1–2 g S8 in 100 g bei RT) auch bei niedriger Temperatur monoklin auskristallisiert, aus dem guten Lösungsmittel Schwefelkohlenstoff (ca. 35 g S8 in 100 g bei RT) aber orthorhombisch.
Ein kurzer Nachtrag: die experimentelle Kontrolle, welche Modifikation vorliegt, gelingt durch Beugungsversuche mit Röntgenstrahlung (die in diesem Skript gezeigten experimentell bestimmten Strukturen wurden so an Einkristallen bestimmt). Zur Charakterisierung im Sinne eines „Fingerabdrucks“ genügt meist ein Beugungsdiagramm, für das anstelle eines einzelnen Kristalls ein Kristallpulver verwendet wird. Die Abbildung zeigt solche „Pulverdiagramme“ für α- und β-S8, die mit Hilfe von Kupfer-Kα-Strahlung aufgenommen wurden; dargestellt ist die Intensität der abgebeugten Strahlung über dem Beugungswinkel 2 θ (das „∼“ kappt den stärksten Reflex der rhombischen Phase):
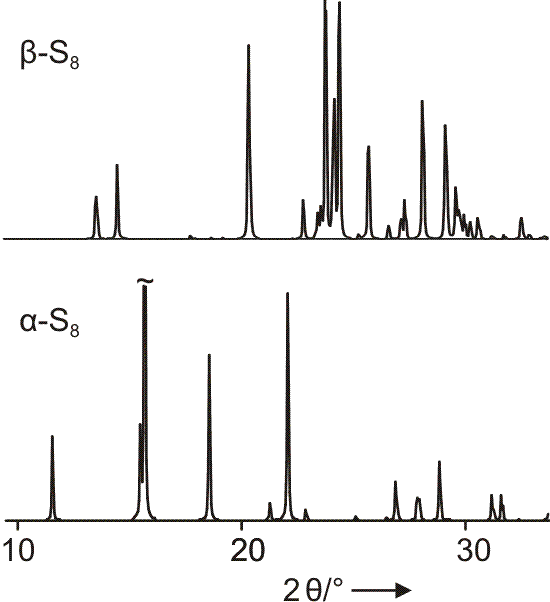
Versuch 22-1: Plastischer Schwefel
Beim Erhitzen von Schwefel über den Schmelzpunkt hinaus kommt es zu Brüchen von S-S-Bindungen und zu deren Neuknüpfung. In Abhängigkeit von der Temperatur bilden sich Ringe anderer Größe und polymere Ketten von Schwefelatomen. Da sich Schwefelringe durch andere Methoden auch gezielt aufbauen lassen, liegen Strukturanalysen an solchen Modifikationen vor. Ein Beispiel ist die Struktur von S20, bei dem der S-S-Atomabstand und der S-S-S-Bindungswinkel den Werten bei S8 entspricht:
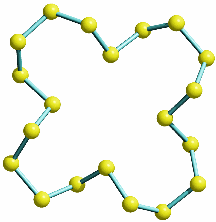
Ein weiteres Strukturelement tritt recht konstant auf: die S-S-S-S-Torsionswinkel weichen nicht weit vom rechten Winkel ab (unter dem Torsionswinkel einer Kette A1-A2-A3-A4 wird der Winkel verstanden, den A1-A2 und A3-A4 einschließen, wenn durch A2-A3 projiziert wird; alternativ: der Winkel zwischen den Ebenen A1-A2-A3 und A2-A3-A4).
Strukturanalysen an polymerem Schwefel, der unter den Bedingungen der Schmelze entstanden ist, fehlen. Ein anschauliches Bild von polymeren Formen des Schwefels vermittelt jedoch die Struktur einer unter Druck entstandenen kristallinen Schwefelmodifikation (Bindungswinkel: 102°):
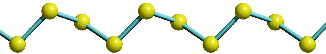
In einer ähnlichen Struktur kristallisiert die stabile Modifikation von Selen:
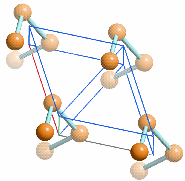
Es zeigt sich hier eine Regel, die im Bereich der Nichtmetall-Grenze generell gilt: die Hochdruckmodifikation eines Elements zeigt die Strukturmerkmale der Normaldruckmodifikation eines schweren (metallischeren) Homologen. Tellur kristallisiert isotyp zur Se-Struktur. An diesem Paar lässt sich der allmählich zunehmende Metallcharakter an einer einzigen Struktur verfolgen. Man vergleiche hierzu die Abstände der beiden kovalent mit denen der sechs nicht-kovalent gebundenen Nachbarn:
| X-X/pm | X···X/pm | |
|---|---|---|
| Se | 2,30 | 4,30 |
| Te | 2,83 | 4,46 |
Eine Hochdruckmodifikation von Tellur zeigt diese Regeln erneut, indem die α-Polonium-Struktur sichtbar wird:
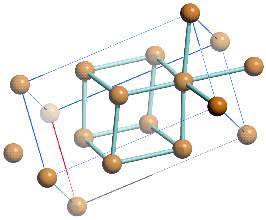
α-Polonium zeigt das einfache kubische Muster mit oktaedrischer Umgebung der Atome unverzerrt:
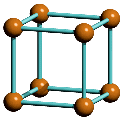
Der Bezug der α-Polonium-Struktur zu einer typischen Metallstruktur, dem A2-Typ, ist im folgenden Bild dargestellt. Um ausgehend von der primitiv-kubischen Struktur zum A2-Typ zu gelangen, ist eine Kompression entlang der langen Raumdiagonalen der rot eingezeichneten Zelle nötig (β-Polonium kristallisiert in einer trigonalen Struktur, die sich auf halbem Weg zum A2-Typ befindet).
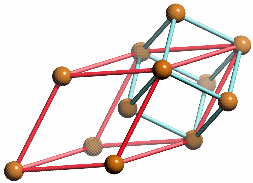
Der deutliche Unterschied zwischen Sauerstoff und Schwefel prägt auch die Chemie der Verbindungen der beiden Elemente.
Versuch 22-5: H2S aus H2 und S
Versuch 22-9: H2S ist eine schwache Säure, S2− ist eine starke Base
Versuch 22-10: Fällung von Metallsulfiden
Die Unterschiede zwischen H2O und H2S beruhen im wesentlichen darauf, dass die freien Elektronenpaare bei H2S keine Lewis- oder Brønsted-Basizität entfalten. Als Folge zeigt Wasser eine Reihe von Eigenschaften und Reaktionen, die von H2S nicht geteilt werden: Wasser wird durch Säuren zu H3O+ protoniert – Wasser ist amphoter, nicht nur Säure; Wasser ist Wasserstoffbrückenbindungs-Akzeptor – nicht nur Donor; Wasser kann wirksam Kationen hydratisieren, es ist ein brauchbarer Ligand und ein gutes Lösungsmittel für polare Stoffe. Alle diese Eigenschaften werden von H2S nicht geteilt, da ihm die Lewis-Basizität hierzu fehlt. Da H2S keine Wasserstoffbrückenbindungen ausbildet, ist es ein Gas, dessen Siedepunkt ca. 130 °C unter dem des Wassers liegt.
Darüber hinaus sind die Molekülstrukturen verschieden. Wassermoleküle zeigen einen H-O-H-Bindungswinkel von 105°, während H2S, H2Se und H2Te Bindungswinkel von 92, 91 und 90° aufweisen.
In welchem Bindungsmodell spiegeln sich diese Unterschiede wider? Das VSEPR-Modell, das für das H2O-Molekül zu einem brauchbaren Ergebnis führte, formuliert in allen Fällen ohne Unterschied vier gleichartige Elektronenpaare – es taugt also nicht, um einen Unterschied zu erklären.
Auch das MO-Schema von H2S ist demjenigen von Wasser sehr ähnlich.
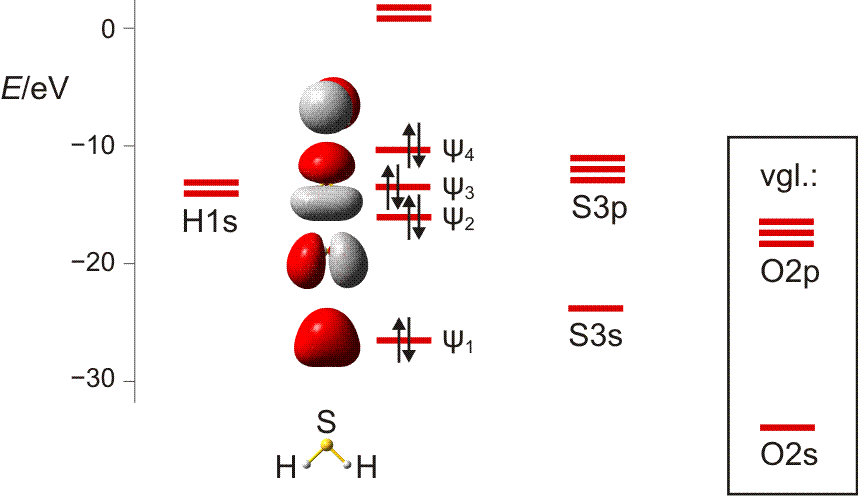
Qualitativ fällt kein Unterschied ins Auge, der s-Anteil der H-S-Bindungen errechnet sich jedoch nur zu 15% – man vergleiche ca. ¼ s-Charakter der H-O-Bindungen im Wassermolekül. Der s-Anteil der freien Elektronenpaare ist also bei H2S größer, bei Gleichverteilung 35% wegen (100% − 2 × 15%) × ½.
Dieses Ergebnis, dass die Eigenschaften von Elementen und deren Verbindungen von der Stellung des Elements innerhalb einer Gruppe des PSE abhängt und dieser experimentelle Befund vor allem mit der unterschiedlichen Beteiligung von s-Orbitalen zusammenhängt, wird im Folgenden vertieft. Eine wesentliche Frage in der Chemie der Nichtmetalle lautet daher: Warum und in welchem Umfang nimmt das s-Orbital der Valenzschale an den Bindungen teil? (Das einfache VSEPR-Modell beantwortet diese Fragen so: Warum? Es ist eben so. In welchem Umfang: vollständig.)
Abschließend eine Anmerkung: da die Diskussion um die Unterschiede der MO- und der VB-Theorie in Teil A schon etwas entfernt sind, werden die dort eingeführten Verfahren und Überlegungen in einer ausführlichen Bindungsdiskussion an H2S und H2O wiederholt.

Die Abnahme der s-Orbital-Beteiligung an den bindenden Elektronenpaaren von H2O zu H2S entspricht einem allgemeinen Trend im Periodensystem. Die s-Orbital-Beteiligung ist bei den leichten Elementen einer Gruppe größer als bei den schweren Homologen, wobei der größte Sprung zwischen der 2. und 3. Periode auftritt. Als Ursache wird oft die abnehmende Übereinstimmung der Bereiche größter Elektronenaufenthaltswahrscheinlichkeit genannt. Während bei 2s- und 2p-Orbitalen die maximale Aufenthaltswahrscheinlichkeit bei ungefähr gleichem Abstand vom Kern vorliegt, ist der s-Zustand bei größerer Hauptquantenzahl zunehmend kernnäher als die entsprechenden p-Orbitale.
Welche Werte ergeben sich konkret und welche Auswirkungen hat der zunehmend kleiner werdende s-Anteil bei den schweren Homologen auf deren Strukturchemie und deren Reaktivität?
Die Rechnungen zeigen hier eine deutliche Abhängigkeit der s-Orbitaltalbeteiligung vom jeweiligen Bindungspartner. Unter den einfachen und grundlegenden Verbindungsklassen werden besonders hohe s-Anteile in Bindungen von Nichtmetallatomen zu zwei Atomsorten gefunden: zu Wasserstoff- und zu Sauerstoffatomen (wirklich O-Atome, nicht OH-Gruppen, etc.!).
Die folgende Tabelle vergleicht die s-Anteile (in %) in Bindungen zwischen einem Nichtmetallatom und einem Wasserstoffatom in den einfachen Element-Wasserstoff-Verbindungen mit dem jeweiligen Element in dessen niedrigster Oxidationsstufe. (Techn. Anm.: mp2/cc-pvtz + nbo.)
| NH3 | 25 | H2O | 24 | HF | 21 | ||
| PH3 | 16 | H2S | 15 | HCl | 15 |
Werden anstelle der Element-Wasserstoff-Bindungen Element-Element-Bindungen betrachtet, so ergeben sich meist kleinere s-Beiträge. Die nächste Tabelle führt Moleküle mit jeweils einer Element-Element-Bindung auf, die außerdem meist noch Wasserstoffatome enthalten. Die nicht tabellierten s-Anteile der Element-Wasserstoff-Bindungen sind in etwa die gleichen wie bei den Verbindungen in der vorigen Tabelle. (Techn. Anm.: mp2/6-311+g(2d,p) + nbo)
| N2H4 | 26 | H2O2 | 12 | F2 | 6 | ||
| P2H4 | 12 | H2S2 | 10 | Cl2 | 7 |
Niedrige s-Anteile in Element-Element-Bindungen bedeuten, dass man sich diese ohne großen Fehler als Bindungen zwischen p-Orbitalen vorstellen kann. Da in diesem Bereich des Periodensystems die p-Orbitale die ungepaarten Elektronen enthalten – die Beteiligung von s-Orbitalen die Betrachtung also komplizierter macht –, stellen Moleküle wie Cl2 oder auch S8 (siehe unten) für die Bindungsbeschreibung kaum gestörte Normalfälle dar. Gedanklichen Aufwand verursacht ein scheinbar unkompliziertes Molekül wie Wasser!
Eine besonders charakteristische Reaktion der Lewis-Base S2− ist der nukleophile Angriff auf S8-Ringe. Diese werden zu Bruchstücken gespalten, wobei Polysulfid-Ionen entstehen. In der folgenden Gleichung steht Sn−1 für Ketten von Schwefelatomen, die durch Spaltung und Neuorganisation im Lösungsgleichgewicht verfügbar sind:
S2− + Sn−1 → Sn2−
Versuch 22-12: Natriumsulfid-Lösung löst Schwefel zu einer gelben Polysulfidlösung
Die in alkalischer Lösung beständigen Polysulfid-Ketten bilden bei Säurezugabe unbeständige Polysulfane H2Sn, die sich unter Schwefelabscheidung zersetzen. Eine charakteristische Eigenschaft der Polysulfide ist deren Oxidierbarkeit. 1-Elektronen-Oxidationsmittel führen leicht zu reaktiven Sn•−-Radikal-Anionen, die sich zu weiteren Schwefel-Polymeren kombinieren können.
Ein besonders ungewöhnliches Mineral ist Lapis lazuli, der in zerriebener Form seit der frühen Antike als Blaupigment Ultramarin geschätzt wird. In der ersten Hälfte des 19. Jahrhunderts gelang die synthetische Herstellung („Ultramarinblau“). Die ungewöhnlichen Eigenschaften des Ultramarins sind in seinem ebenso ungewöhnlichen Aufbau begründet. In einem offenen Silicatnetzwerk, einem „Zeolith“ (siehe das Kapitel über die 14. Hauptgruppe), sind enge Hohlräume vor allem durch S3•−-Radikal-Anionen gefüllt. Die Fixierung in der Silicatstruktur verhindert die üblichen Reaktionen dieses reaktiven Radikals, an dessen elektronischem Zustand die gezeichneten mesomeren Grenzformeln ungefähr den gleichen Anteil haben.
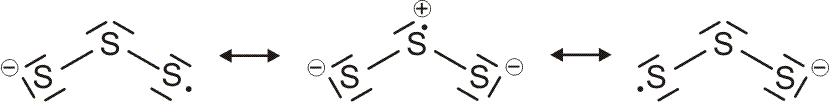
Versuch 22-13: Ultramarinblau und Lapis lazuli zeigen
Schwefel verbrennt in einer exothermen Reaktion zu Schwefeldioxid (ΔfH° = −297 kJ mol−1):

Man beachte die Formulierung, die im Einklang mit der Oktettregel erfolgt. Die Rechnung zeigt, dass die S-O-σ-Bindungen ca. 17 % s-Charakter aufweisen, das freie Elektronenpaar hat nahezu 70 % s-Charakter. Auch hier zeigt sich der typische Unterschied zwischen einem Element der zweiten und der dritten Periode: Ozon wird durch analoge mesomere Grenzformen beschrieben, die s-Anteile in den Bindungen und dem freien Elektronenpaar am mittleren O-Atom sind jedoch charakteristisch: 24 % in den Bindungen, 52 % beim freien Elektronenpaar.
Versuch 22-3c: Verbrennung von Schwefel
• S + O2 → SO2
SO2 ist ein Säureanhydrid. Mit Wasser wird schweflige Säure gebildet:
SO2 + H2O → H2SO3
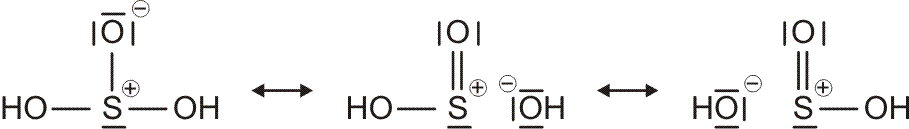
Bei der Formulierung der Lewis-Formeln wird auf ein Problem eingegangen, das durch die Absicht entsteht, die Oktettregel einzuhalten. Die linke mesomere Grenzformel zeigt das H2SO3-Molekül in der zum Beispiel bei Mortimer üblichen Weise. Hierbei haben alle Bindungen die kovalente Bindungsordnung 1, es wird also nicht zwischen der Bindung zur OH-Funktion und der zum O-Atom unterschieden. Hier ist die (zu vermeidende) hypervalente Formulierung mit 10 Elektronen hilfreicher, bei der durch eine Doppelbindung zum O-Atom hervorgehoben wird, dass diese Bindung die doppelte Bindungsordnung der S–OH-Bindungen hat. Eine realistischere Beschreibung des Moleküls unter Erhalt der Oktetts ist jedoch möglich, indem die beiden rechten mesomeren Grenzformeln hinzugenommen werden. Beachten Sie, dass diese Betrachtung auch auf weitere wichtige Moleküle zutrifft. Beispiele sind Schwefelsäure und Phosphorsäure.
Schweflige Säure zeigt eine weitere Besonderheit, indem sie in der beschriebenen SO(OH)2-Form als schweflige Säure im engeren Sinne auftritt, diese aber im Gleichgewicht mit „Sulfonsäure“, HS(O)2(OH), steht. Wir werden Einzelheiten dazu erst später behandeln und zwar beim Gleichgewicht zwischen „phosphoriger Säure“ und Phosphonsäure.
Schweflige Säure ist aufgrund ihrer hohen Tendenz, zu Sulfat oxidiert zu werden, ein gebräuchliches Reduktionsmittel.
Versuch 22-20: SO2 reduziert Manganat(VII)
• 2 MnO4− + 5 SO2 + 6 H2O → 2 Mn2+ + 5 SO42− + 4 H3O+
Versuch 22-21: SO2 reduziert Iod
• SO2 + I2 + 6 H2O → SO42− + 2 I− + 4 H3O+
Versuch 22-23: SO2 reduziert selenige Säure
• 2 SO2 + H2SeO3 + 5 H2O → 2 SO42− + Se + 4 H3O+
In der Technik entsteht Schwefeldioxid bei der Verbrennung schwefelhaltiger Verbindungen. Da Schwefeldioxid ein Luftschadstoff ist, wird er in Kraftwerksanlagen dem Abgasstrom entzogen. Bei dieser Rauchgasentschwefelung entsteht nach der Oxidation zu Sulfat und der Reaktion mit Löschkalk, Ca(OH)2, der sogenannte Chemiegips, dessen weitere Nutzung ein Problem darstellt.
Versuch 22-22: SO2-Absorption mit Löschkalk
Schwefelsäure, H2SO4 ist aufgrund ihrer Verwendung bei der Düngemittelherstellung mengenmäßig eine der bedeutendsten Grundchemikalien (vgl. bei der 15. Hauptgruppe, Phosphatdünger). Beim Kontaktverfahren wird Schwefeldioxid katalytisch mit Sauerstoff zu Schwefeltrioxid oxidiert.
Versuch 22-26: Kontaktverfahren zur Herstellung von Schwefelsäure
Versuch 22-27a: SO3 als Schwefelsäureanhydrid
Das entstehende Schwefeltrioxid fällt bei der Herstellung als Gas an, bei Raumtemperatur bildet es jedoch ein farbloses festes Polymer oder in einer weiteren Modifikation ein Trimer:
Die Bildung von Schwefeltrioxid aus Schwefeldioxid verläuft ebenfalls exotherm:
SO2 + ½ O2 ⇄ SO3 ΔrH° = −98 kJ mol−1
Es stellt sich die Frage, warum beim Verbrennen von Schwefel nicht gleich die exotherme Gesamtreaktion mit Schwefeltrioxid als Produkt abläuft (wir reden darüber, überlegen Sie schon mal).
Schwefeltrioxid bildet als Säureanhydrid mit Wasser Schwefelsäure. Im technischen Prozess wird jedoch nicht diese Reaktion eingesetzt, sondern SO3 wird in konzentrierte Schwefelsäure eingeleitet, mit der das Säureanhydrid „Oleum“ bildet, einem Gemisch von Schwefelsäure und Isopolysäuren des Schwefels. Deren einfachster Vertreter ist die Dischwefelsäure:
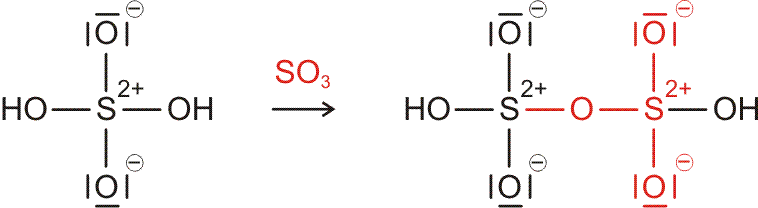
Wird eine zweite SO3-Einheit in eine S-OH-Funktion inseriert, entsteht „Trischwefelsäure“.
Schwefelsäure gehört zu den am häufigsten eingesetzten Chemikalien, zugleich ist ihre hohe Reaktivität und Aggressivität zu beachten.
Versuch 22-28: Sichere Handhabung von Schwefelsäure
Versuch 22-29: Konzentrierte Schwefelsäure zerstört organische Verbindungen
Versuch 22-30: Schwefelsäure oxidiert Kupfer
• Cu + H2SO4 + 2 H3O+ → Cu2+ + SO2 + 4 H2O
Versuch 22-31a: Schwefelsäure oxidiert Iodid
• 2 I− + H2SO4 + 2 H3O+ → I2 + SO2 + 4 H2O
Versuch 22-31b: Schwefelsäure oxidiert Zink
• 4 Zn + H2SO4 + 8 H3O+ → 4 Zn2+ + H2S + 12 H2O, daneben auch:
• 3 Zn + H2SO4 + 6 H3O+ → 3 Zn2+ + S + 10 H2O
Versuch 22-24: Bildung von Zinkdithionit
Versuch 22-32: Natriumthiosulfat aus Natriumsulfit und Schwefel; Zersetzung von Thioschwefelsäure
Versuch 22-33: Thiosulfat als Ligand für Silber(I)
Versuch 22-34: Thiosulfat als Schwefelquelle
Versuch 22-35: Iod und Thiosulfat
Die Reaktion von Sulfid mit Schwefel unter Bildung von Schwefel-Schwefel-Bindungen ist eine besonders typische Umsetzung, da ein analoges Verhalten auch mit anderen Oxidationsstufen des Schwefels beobachtet wird. Schwefel-Schwefel-Bindungen können darüber hinaus bei Redoxreaktionen gebildet werden. So zeigen die folgenden Versuche zuerst die reduktive Kupplung von Sulfit-Ionen zu Dithionit:
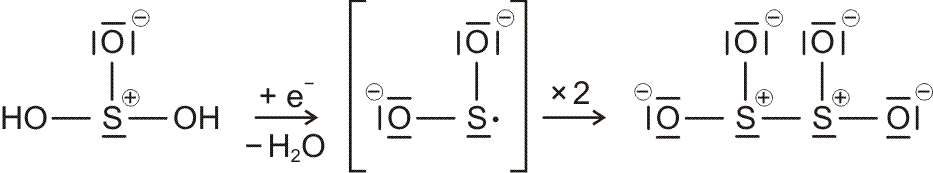
anschließend dann die Umsetzung von Sulfit mit Schwefel zu Thiosulfat:
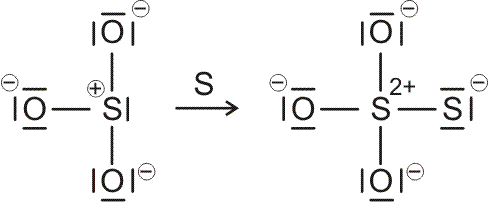
Dessen Umsetzung mit Iod führt schließlich durch oxidative Kupplung zu einem Salz einer Polythionsäure und zwar zu Tetrathionat:
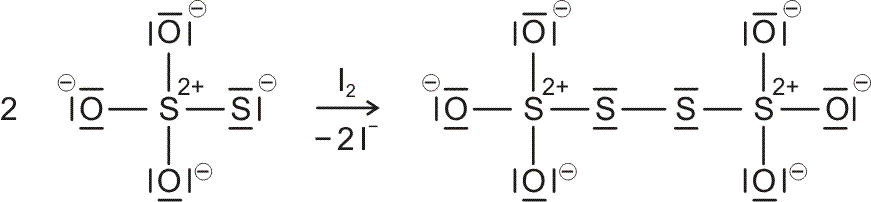
Die Bildung von Schwefel-Schwefel-Bindungen führt zu Strukturelementen, die bei den Schwefelmodifikationen bereits sichtbar waren: Schwefelatomketten mit S-S-Abständen etwas oberhalb von 200 pm und Bindungswinkeln etwas oberhalb von 100°. Auffallend beim Tetrathionat vor allem wieder der fast rechte S-S-S-S-Torsionswinkel (abgebildet ist die Struktur des Anions in monoklinen Kristallen von K2S4O6):
Wie lässt sich dieses wiederkehrende Strukturelement verstehen? Hierzu zunächst ein Blick auf Schwefeldichlorid.
Schwefeldichlorid ist eine reaktive Verbindung, die technisch als Grundchemikalie hergestellt und in der Regel sofort weiterverarbeitet wird (SCl2 ist instabil gegen den Zerfall in Chlor und S2Cl2). SCl2 besteht aus gewinkelten Molekülen, die im Einklang mit der Oktettregel formuliert werden können:
Die Umgebung des Schwefelatoms im SCl2-Molekül ist derjenigen in Schwefelketten oder -ringen sehr ähnlich: der Abstand zu den Chlor-Nachbarn ist etwas größer als 200 pm (206 pm) und der Bindungswinkel ist etwas größer als 100° (104°). SCl2 gehört zu den Molekülen, für deren Formulierung sich nur eine einzige Lewis-Formel anbietet. Die Beschreibung durch lokalisierte Bindungen ist daher angemessen. Auch hier ist wenig Aufregendes zu finden; das folgende Bild zeigt eine der beiden gleichwertigen S-Cl-Bindungen:
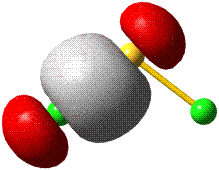
Die dargestellte Bindung ist im Wesentlichen durch die Wechselwirkung eines S(3p)- und eines Cl(3p)-Orbitals entstanden. Der s-Anteil ist bei beiden Atomen klein; beim Schwefelatom wird ein Beitrag von 94 % p-Charakter nur durch 6 % s-Anteil modifiziert. Dementsprechend sind die beiden freien Elektronenpaare zu formulieren. Senkrecht auf der Ebene der drei Atome steht ein reines p-Orbital als eines der beiden freien Schwefel-Elektronenpaare. In der Ebene ist vom s-Orbital zweimal der 6-%-ige Beitrag zu den Bindungen abzuziehen, für das freie Elektronenpaar ergibt sich also 12 % p-Anteil und 88 % s-Charakter. Kurz: Vernachlässigen Sie im SCl2 jegliche Hybridisierung. SCl2 ist damit ein Molekül, dessen Bindungen sich sofort aus den beteiligten Atomen in deren Grundzustand ergeben.
Wie aber passt diese Aussage zum Bindungswinkel von 104°? Wir hatten bei Wasser und Schwefelwasserstoff gelernt, auf die Abstoßung zwischen Elektronenpaaren zu achten. Wird SCl2 hierauf untersucht, so fällt eine Gruppe von Orbitalen auf, deren Platzbedarf den erwarteten 90°-Bindungswinkel wirksam verhindert: die freien Elektronenpaare an den Chlor-Atomen und hier besonders die Elektronenpaare in p-Orbitalen. Die folgende Abbildung zeigt ein solches Orbital:
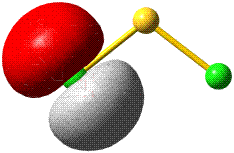
Es wird deutlich, dass sich das abgebildete Elektronenpaar und dessen Gegenüber am anderen Chloratom den Raum aufteilen müssen. Eine Verkleinerung des Cl-S-Cl-Winkels würde zu vermehrter Pauli-Abstoßung führen, so dass der beobachtete 104°-Winkel die Balance darstellt zwischen dieser Abstoßung und dem Gewinn an Bindungsstärke bei optimaler Überlappung der Bindungsorbitale.
Auf diese einfache Weise erschließt sich auch die Strukturchemie der Schwefelketten und -ringe. In der folgenden Bilderserie ist dargestellt (von links): ein S8-Molekül mit Kronenstruktur, bei der vier Atome näher zum Betrachter hin liegen und vier darunter. Links daneben ist eine S-S-Bindung dargestellt, die völlig derjenigen im SCl2 entspricht (9 % s- und 90 % p-Anteil). Dementsprechend zerfallen die beiden freien Elektronenpaare an jedem Schwefelatom in ein Orbital, das im Wesentlichen s-Charakter aufweist und in ein reines p-Orbital, das senkrecht auf der Ebene steht, die neben diesem Schwefelatom dessen beiden nächsten Nachbarn enthält. In den beiden rechten Bilder sind zwei dieser Orbitale dargestellt. Es wird deutlich, dass die Abstoßung der freien p-Elektronenpaare minimiert wird, wenn diese im rechten Winkel zueinander stehen. Die Abstoßung zwischen freien p-Elektronenpaaren ergibt sich damit als die einfache Ursache für das häufige Auftreten von 90°-Torsionswinkeln.
Es wird erneut deutlich, welch komplizierter Sonderfall ein Wassermolekül darstellt, bei dem mehr Einflussgrößen als bei SCl2 und bei den Schwefelketten und -ringen in der realen Struktur ausbalanciert sind.
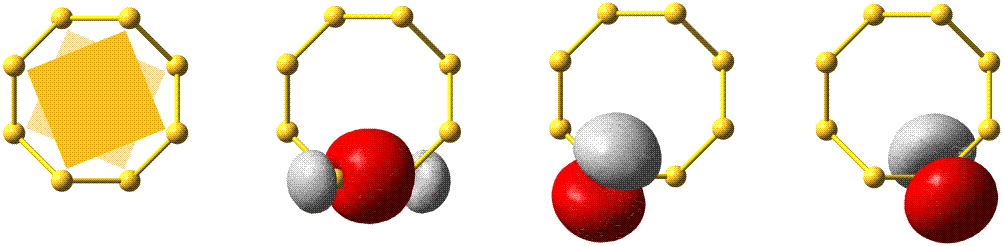
Während bei Sauerstoff nur die dem H2O und dem H2O2 formal entsprechenden Fluoride OF2 und O2F2 bekannt sind, bildet Schwefel auch Fluoride in höheren Oxidationsstufen aus.
Die ungewöhnlichste Verbindung ist wohl das farblose Gas Schwefelhexafluorid, SF6. Während die Fluoride von Edelgasen und Halogenen – aber auch das näher verwandte SF4 – reaktive Fluorierungsreagenzien darstellen, ist SF6 unreaktiv: obwohl sich ein sehr deutlich negativer ΔG-Wert für die Spaltung der sechs S-F-Bindungen bei der Reaktion mit Wasser ergibt, findet selbst mit Wasserdampf von 500 °C keine Reaktion statt. SF6 wird wegen dieses Verhaltens technisch genutzt, zum Beispiel als unzersetzliches und elektrisch isolierendes Schutzgas in der Hochspannungstechnik. Die thermodynamischen Kennzahlen zeigen, dass SF6 keine besondere Stabilität aufweist. SF6 ist vielmehr inert, seine Unreaktivität ist auf ungewohnlich hohe Aktivierungsenergien für die Umsetzung mit Reaktionspartnern jeglicher Art zurückzuführen. Die mangelnde Reaktivität scheint mit der sterischen Abschirmung des Schwefelatoms durch sechs Fluoratome zusammenzuhängen:
Beachten Sie die unterschiedlichen Begriffswelten: Ist eine Verbindung oder eine Reaktionsmischung in einem Zustand, der einem Energieminimum in Bezug auf alle denkbaren Zerfalls- und Reaktionsprodukte entspricht, so sind sie stabil; wenn nicht, sind sie unstabil. Sind eine Verbindung oder eine Reaktionsmischung unstabil, werden aber durch hohe Aktivierungsbarrieren an der Reaktion gehindert, so sind sie inert; wenn nicht, sind sie labil. Die Begriffspaare stabil/unstabil und inert/labil sind vollständig unabhängig voneinander. Die oben behandelten Protolysegleichgewichte in wässriger Lösung stellen sich augenblicklich ein, ohne erkennbare Aktivierung: nach der Gleichgewichtseinstellung sind solche Lösungen demnach stabil und labil. Das Stickstoff/Sauerstoff-Gemisch Luft ist gegenüber der Reaktion zu Stickstoffoxiden stabil und inert (Gott sei Dank). Stickstoffmonoxid, NO, ist bei Raumtemperatur unstabil und inert. Es ist didaktisch nicht besonders toll, aber sehr weit verbreitet, diese Kombination metastabil zu nennen, obwohl die getrennten Welten der Thermodynamik und der Kinetik in diesem Begriff vermischt werden. (Darüberhinaus ist es schade, dass Sie in manchen Büchern statt inert/labil die Begriffe „kinetisch stabil/unstabil“ finden.)
SF6 ist bedeutend genug, um an diesem Beispiel die Behandlung der Bindung in Molekülen fortzusetzen. Bisher war ein Fall nicht besprochen worden: wie werden Moleküle beschrieben, die bei der Annahme von 2e-2z-Bindungen das Valenzelektronenoktett überschreiten, bei denen das Zentralelement aber in der höchstmöglichen Oxidationsstufe vorliegt? Man erinnere sich an eine Vereinfachung, die bei Molekülen angewandt wurde, bei denen das Zentralelement noch mindestens 1 freies Elektronenpaar aufwies: diese Elektronen wurden dem s-Orbital zugewiesen und die Bindungen dann nur mit p-Orbitalen formuliert. Sind alle Valenzelektronen an der Verbindungsbildung beteiligt – was zum Beispiel der Fall ist, wenn die Oxidationsstufe der Zahl der Valenzelektronen entspricht – hat diese Vereinfachung natürlich keine Grundlage mehr.
In diesem Fall ist die Strukturvorhersage unaufwendig, dafür erfordert es mehr Mühe, die Bindungen im Einklang mit der Oktettregel zu beschreiben. Gehen Sie folgendermaßen vor:
• (zuerst wieder zur Sicherheit:) Liegt das Zentralelement in der höchsten Oxidationsstufe vor? Bei SF6 ist die Oxidationsstufe des Schwefels +VI, bei einem Element mit 6 Valenzelektronen also ja.
• Die Substituenten bilden in diesem Fall ein reguläres Polyeder um das Zentralatom und erreichen damit den größtmöglichen Abstand voneinander. Bei 4 Substituenten ist dies ein Tetraeder, bei 5 eine trigonale Bipyramide, bei 6 ein Oktaeder. Die Struktur von SF6 ist also oktaedrisch.
• Stelle den Beitrag der p-Orbitale des Zentralatoms fest: markiere hierzu lineare B-A-B-Einheiten, in denen 4e-3z-Bindungen auftreten und ordne jeder Bindung die kovalente Bindungsordnung ½ zu. Bei SF6 betrifft dies alle sechs Bindungen; jede erhält einen p-Beitrag zur Bindungsordnung von ½.
• Addiere den Beitrag des s-Orbitals des Zentralatoms: in einem regulären Polyeder überlappt das s-Orbital gleichwertig mit den Orbitalen aller Substituenten. Bei n Substituenten trägt diese Wechselwirkung zu 1/n zu jeder Bindung bei (Diese Wechselwirkung ist zum Beispiel im „Mortimer“ in Abb. 9.25 dargestellt, im „Riedel“ in Abb. 2.75a). Bei SF6 ist dieser Beitrag 1/6, die gesamte kovalente Bindungsordnung ist die Summe von s- und p-Anteil, also 2/3. Die Oktettregel ist damit erfüllt (6 × 2/3 × 2 Elektronen = 8 Elektronen).
Auf die bei SF6 angewandte Weise werden weitere Verbindungen mit scheinbarer Überschreitung des Elektronenoktetts behandelt. Eine für die weiteren Überlegungen wichtige Verbindung ist die Tellursäure H6TeO6 = Te(OH)6. Man beachte, dass hier gegenüber der Schwefelsäure eine formal wasserreichere Form mit erhöhter Koordinationszahl vorliegt.
Die Bedeutung von Elektronenabzählregeln lässt sich sehr unmittelbar in der Chemie der Chalkogen-Kationen beobachten. Solche Kationen entstehen durch Oxidation von Schwefel, Selen und Tellur durch Oleum:
Versuch 22-4: Schwefel, Selen und Tellur in Oleum
Durch Zusatz geeigneter Gegenionen lassen sich Salze mit den oxidierten Formen der drei Elemente bilden und strukturell untersuchen. Durch die Oxidation sind Kationen entstanden, die sich vom Element nur durch Elektronenentzug ableiten. So entsteht auch das Te82+-Ion, dem zwei Elektronen gegenüber dem Element fehlen. Die Strukturanalyse zeigt die Auswirkung des Elektronenmangels: die beiden Radikale an zwei oxidierten Positionen bilden eine kovalente Bindung quer über eine Te8-Ring und stellen damit die gewünschte Elektronenbilanz wieder her:
Schwefel reagiert in analoger Weise, ohne jedoch strukturell so deutlich wie Tellur eine kovalente Einfachbindung mit normalem S-S-Abstand aufzubauen:
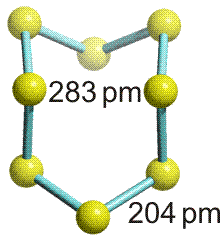
Einfache Modellvorstellungen legen nahe, dass jedes Molekül mit einem freien Elektronenpaar an einem der Atome basische Eigenschaften haben müsste. Dabei sind Neutralbasen eine echte Rarität. Da gibt es Ammoniak und seine Derivate, außerdem … so gut wie nichts. Wir schauen in diesem Kapitel Orbitale an, um das zu verstehen.
Stickstoff kommt in seinen Verbindungen in allen denkbaren Oxidationsstufen von N − 8 bis N (N = Zahl der Valenzelektronen), also von −III bis +V vor. Die stabilste Form des Stickstoffs ist das N2-Molekül, der sowohl für die technische Nutzung als auch durch die belebte Natur aktiviert wird, indem N2 zuerst zur Oxidationsstufe −III reduziert wird – technisch durch das Haber-Bosch-Verfahren, in der Natur durch Bakterien der Gattung Rizobium, die mit verschiedenen Arten aus der Familie der Fabaceae („Leguminosen“, Schmetterlingsblütler), zum Beispiel mit Lupinen, in Symbiose leben.
Ammoniak wird durch stickstoffassimilierende Bakterien aus N2 gebildet, das im aktiven Zentrum des Enzymkomplexes Nitrogenase zur Oxidationsstufe −III reduziert wird. Die Bildung von Stickstoff(−III) und die anschließende Protonierung eines N3− (Nitrid) zu Ammoniak findet unter erheblich drastischeren Bedingungen in einem einfachen Schulversuch statt.
Versuch 23.1.2: Magnesium bildet beim Verbrennen an der Luft bei Sauerstoffmangel auch Nitrid, das mit Wasser zu Ammoniak reagiert
• 3 Mg + N2 → Mg3N2
• Mg3N2 + 6 H2O → 3 Mg(OH)2 + 2 NH3
Die folgenden Versuche zeigen eine Reihe von Eigenschaften des Ammoniaks, die anschließend durch die besondere Bindungssituation im NH3-Molekül gedeutet wird: NH3 ist wasserähnlich, aber doch ein Gas; NH3 ist – obwohl Neutralmolekül – eine Base:
Versuch 23.1.6: NH3-Springbrunnen • ähnlich wie bei HCl ist bei NH3 die Löslichkeit in Wasser extrem: auch Ammoniak kann Wasserstoffbrückenbindungen ausbilden; beim Auflösen findet in geringem Umfang (pKB = 4,8) Protolyse statt.
Versuch 23.1.7: NH3 als Base: Salzbildung mit HCl
Versuch 23.1.3: NH4Cl reagiert mit Natronlauge zu NaCl und Ammoniak
Als Base bildet NH3 Salze, in denen das Ammonium-Kation, NH4+, vorliegt. Die auch bei der ionischen Bindung gefundene homolytische Bindungsspaltung bei Zufuhr der Bindungsdissoziationsenergie wird bei Ammoniumsalzen in einer Variante gefunden, die demselben Ziel dient, nämlich eine energetisch ungünstige Ladungstrennung zu vermeiden.
Versuch 23.1.8: Ammoniumchlorid sublimiert unter HCl- und NH3-Bildung
Bildet die schwache Base Ammoniak mit schwachen Säuren Salze, so sind diese oft hinsichtlich Aufbau und Reaktivität ungewöhnlich. Beispiele sind die unbeständigen Ammoniumsalze der Kohlensäure, die durch Wasserabspaltung Amidbindungen aufbauen, die durch katalysierte (Harnstoff) und unkatalysierte Hydrolyse (Carbaminat, auch: Carbamat) wieder in Ammoniak und Kohlensäure übergehen. Die Formeln stellen die Hydrolyse ausgehend von Harnstoff dar:
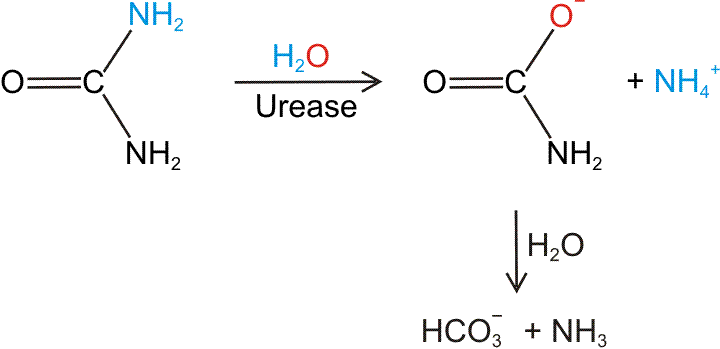
Versuch 23.1.9: Ammoniumcarbaminat
Ein weiteres reaktives Ammoniumsalz ist „Ammoniumnitrit“ – in Anführungsstrichen, da dessen Beständigkeit zu gering ist, als dass das isolierte Salz bekannt wäre. Die Zersetzung findet bereits statt, wenn Ammonium- und Nitrit-Ionen in erwärmter wässriger Lösung zusammentreffen. Die Reaktion ist historisch von Bedeutung: da der so synthetisierte Stickstoff eine geringere Dichte besitzt als Luft, die von Sauerstoff, Wasser und Kohlendioxid befreit worden ist, wurde die Existenz der Edelgase erkannt.
Versuch 23.1.1: Zersetzung von Ammoniumnitrit
• NH4+ + NO2− → N2 + 2 H2O
Die beiden letzten Versuche heben noch einmal auf die Lewis-Basizität des Ammoniaks ab, durch die es zum Ligand wird, nämlich für Kupfer(II), aus dessen Hydrathülle Wasserliganden verdrängt werden, aber auch für Natrium-Kationen in den ungewöhnlichen Lösungen des elementaren Natriums in flüssigem Ammoniak, die sich bei Aktivierung unter Wasserstoffentwicklung und Natriumamid-Bildung (NaNH2) zersetzen können.
Versuch 23.1.10: Ammoniak als Ligand für Kupfer(II)
• [Cu(H2O)6]2+ + 4 NH3 → [Cu(NH3)4(H2O)2]2+ + 4 H2O
Versuch 23.1.11: Natrium löst sich in flüssigem Ammoniak
• Na + n NH3 → [Na(NH3)n]+ + e−solv
Die nun zusammengetragenen Eigenschaften des Ammoniaks sind recht ungewöhnlich. Sie treten zum Teil nur bei Ammoniak und seinen nächsten Verwandten auf, zum Beispiel die Eigenschaft Neutralbase zu sein. Die Sonderstellung des Ammoniaks wird im direkten Vergleich mit seinem schweren Homologen Phosphan („Monophosphan“), PH3, besonders deutlich. Während Aufbau und Eigenschaften der den Nitriden entsprechenden Phosphide noch einige Gemeinsamkeiten aufweisen, zum Beispiel die Hydrolyseempfindlicheit, fehlt dem Monophosphan die Basizität weitgehend.
Versuch 23.2.8a: Phosphan aus Calciumphosphid
• Ca3P2 + 6 H2O → 3 Ca(OH)2 + 2 PH3
Versuch 23.2.8b: Phosphoniumiodid und Wasser
• PH4+I → PH3 + HI
Werden organische Derivate in die Betrachtung einbezogen, so ergeben sich weitere Unterschiede: Bei den Molekülen von Triorganylphosphanen mit drei verschiedenen Substituenten PR1R2R3 unterscheiden sich Bild und Spiegelbild. Diese Phosphane sind chiral. Geeignete Trennverfahren erlauben es, die beiden Formen, die Enantiomere in die chiralen Reinstoffe zu zerlegen. Chirale Phosphane sind zum Beispiel bei der katalytischen Synthese chiraler Wirkstoffe von Bedeutung.
Chirale Amine des einfachen Typs NR1R2R3 lassen sich dagegen nicht in Form reiner Enantiomere gewinnen und nutzen.
Werden die beim Paar H2O/H2S erkannten Regeln auf NH3 und PH3 angewandt, so werden alle Eigenheiten des Ammoniaks klar:
N-H-Bindungen weisen ca. 25 % s- und 75 % p-Anteil auf. Die drei N-H-Bindungen im Ammoniak lassen daher für das freie Elektronenpaar am Stickstoffatom nur dasselbe s:p-Verhältnis zu. Im Ammoniakmolekül liegen daher alle vier Elektronenpaare in sp3-Hybridisierung vor. (Unter den einfachen Verbindungen dürfte Ammoniak damit das einzige Molekül mit einem freien Elektronenpaar sein, für welches das VSEPR-Modell gilt.) Das freie Elektronenpaar liegt damit bereits im Ammoniak in der Hybridisierung vor, die es in einer Bindung, zum Beispiel im Ammonium-Ion, auch aufweisen würde. Die Basizität, also die leichte Protonierbarkeit des Ammoniaks zum Ammonium-Ion, geht also mit einer Übereinstimmung des elektronischen Zustands von Base und konjugierter Säure einher.
Bei Monophosphanen ist das anders: P-H-Bindungen haben einen s-Anteil von nur ca. 1/6, so dass das freie Elektronenpaar einen erheblich höheren s-Anteil aufweist als beim Ammoniak: ca ½ s- und ½ p-Anteil. Nicht nur die bei Monophosphan stark zurückgedrängte Basizität, sondern auch der Stabilitätsunterschied unsymmetrisch substituierter Phosphane und Amine sind eine direkte Folge dieser Unterschiede in der Bindung. Die folgende Abbildung zeigt die maßgeblichen Orbitale (Ammoniak oben, Monophosphan unten; links die beiden Moleküle, in der Mitte eines der drei Bindungselektronenpaare, rechts das freie Elektronenpaar); man beachte die Ähnlichkeit von Bindungselektronenpaaren und freiem Elektronenpaar bei Ammoniak:
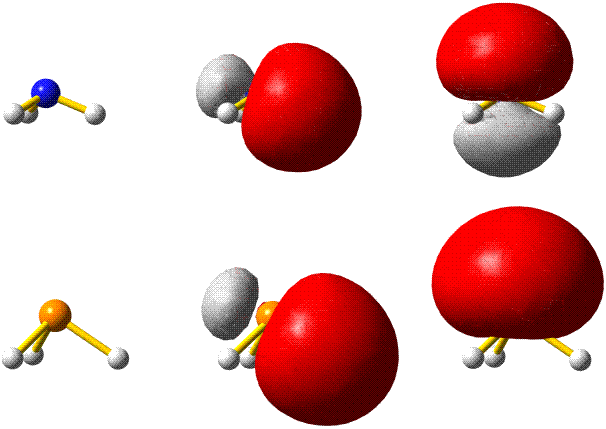
Der Unterschied der Bindungswinkel zwischen NH3 und PH3 ist ungefähr der Gleiche wie beim Paar H2O/H2S (NH3 107°, PH3 93°) und lässt sich auf dieselbe Weise als Zusammenspiel der unterschiedlichen Hybridisierung und der Abstoßung der Elektronenpaare interpretieren.
Der untypisch hohe p-Anteil des freien Elektronenpaars am Stickstoffatom hat neben der Basizität des Ammoniaks eine weitere Konsequenz. Das Fehlen isolierbarer Enantiomere bei einem Amin wie N-Methylanilin (N-Methyl-phenylamin) geht auf eine ungewöhnliche Eigenschaft der Stammverbindung NH3 zurück. Dessen Konfiguration ist nämlich nicht stabil, sondern das Stickstoffatom schwingt andauernd durch die Ebene der drei Wasserstoffatome hindurch. Monophosphan zeigt diese Erscheinung nicht. Die beiden stabilen Anordnungen an den Endpunkten der Schwingung – die trigonal-pyramidale Struktur des Ammoniaks – sind durch einen unstabilen trigonal-planaren Übergangszustand miteinander verbunden. Die zu überwindende Energiedifferenz ist jedoch so klein, dass die Schwingung bei Raumtemperatur stattfindet, während sie bei Monophosphan so groß ist, dass die Konfiguration stabil ist. Der Übergangszustand ist im folgenden Bild dargestellt:
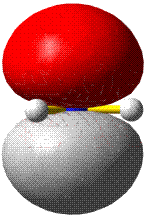
Das Stickstoffatom liegt hier in sp2-hybridisierter Form vor, das freie Elektronenpaar besetzt ein reines p-Orbital. Der Übergangszustand ist damit bei Ammoniak dem Grundzustand erheblich ähnlicher als dies bei Monophosphan der Fall ist, entsprechend gering ist die energetische Barriere bei NH3.
Neben Ammoniak sind weitere Verbindungen mit N-H-Bindungen bekannt. Bedeutung haben Hydrazin, N2H4, und Hydroxylamin, NH2OH. Beide sind aufgrund ihrer Neigung, zu Stickstoff oxidiert zu werden, starke Reduktionsmittel.
Versuch 23.1.16: Reduktion von Kupfer(II)-acetat durch Hydrazin
• 2 Cu2+ + N2H4 + 4 OH− → 2 Cu + N2 + 4 H2O
Eine im Mittel gebrochene Oxidationsstufe liegt in der Stickstoffwasserstoffsäure, HN3, vor, in der ein fast lineares N3-Fragment auftritt. Für HN3 können mehrere Lewis-Formeln gezeichnet werden, die im Einklang mit der Oktettregel sind; diejenige mit dem größten Gewicht ist wohl
HN3 ist eine schwache Säure, die mit einer Vielzahl von Metallen Azide bildet, die zum Teil sehr leicht unter Stickstoffentwicklung zerfallen.
Versuch 23.1.17b: Zersetzung von Blei(II)-azid.
Anwendung findet die Azidzersetzung in Airbags (NaN3). Azid wird zu den Pseudohalogenid-Ionen gezählt, da es in seinen Eigenschaften an die Halogenide erinnert.
Wird die HN-Funktion in HN3 durch das isoelektronische Sauerstoffatom ersetzt, wird Distickstoffmonoxid, N2O („Lachgas“) erhalten, das für Narkosezwecke, als Oxidationsmittel anstelle von Sauerstoff und – im Alltag – als Treibgas für die Sahnebereitung eingesetzt wird. Neben Kohlendioxid und Methan ist Lachgas das dritte bedeutende Treibhausgas in unserer Atmosphäre. Auch bei N2O können wieder mehrere mesomere Grenzformeln im Einklang mit der Oktettregel formuliert werden, das wohl größte Gewicht hat die Grenzformel
Versuch 23.1.18a: Herstellung von Lachgas aus Ammoniumnitrat
• NH4NO3 → N2O + 2 H2O
Der hier gezeigten kontrollierten Zersetzung von Ammoniumnitrat bei relativ niedriger Temperatur steht der stark exotherm verlaufende Zerfall unter Stickstoffbildung bei höherer Temperatur gegenüber, der mehrmals zu verhehrenden Unfällen geführt hat. Bei dem ersten und zugleich größten Zwischenfall dieser Art kamen am 21. September 1921 im dabei vollständig zerstörten Ludwigshafener Stadtteil Oppau mehr als 500 Menschen ums Leben, als eine Menge von 4.500 t Ammoniumnitrat bei dem Versuch explodierte, das festgebackene hygroskopische Düngemittel mit Hilfe von Dynamit wieder in handhabbare Portionen zu zerteilen.
Versuch (neu): Ein weiterer Versuch mit gefährlicher Historie: der bellende Hund
Bei der Reaktion von Stickstoffdioxid mit Wasser entsteht in Anwesenheit von Sauerstoff Salpetersäure, in Abwesenheit von O2 Salpetersäure und salpetrige Säure, wobei letztere in saurer Lösung unbeständig ist und zu Salpetersäure und NO zerfällt.
Versuch 23.1.25: Disproportionierung von NO2 in Wasser
• NO2 + H2O → HNO2 + HNO3
• 3 HNO2 → 2 NO + HNO3 + H2O
Versuch 23.1.32: Unbeständigkeit von HNO2 im Sauren
Man beachte bei der Formulierung der Lewis-Formeln die Oktettregel; für Salpetersäure:
und für salpetrige Säure:
Für die konjugierten Basen, die Nitrite und Nitrate, ergibt sich jeweils eine mesomere Grenzformel mehr als bei den zugrunde liegenden Säuren.
Alkalinitrate spalten beim Erhitzen Sauerstoff ab und gehen dabei in Nitrite über.
Versuch 23.1.27: Erhitzen von Natriumnitrat
• NaNO3 → NaNO2 + ½ O2
Beide Salze können durch eine charakteristische Reaktion nachwiesen werden, die auf dem Zerfall von salpetriger Säure im Sauren und der anschließenden Bindung des Zerfallsprodukts NO an Eisen(II) beruht. Dabei wird das Komplex-Kation [Fe(H2O)5(NO)]2+ gebildet, das näherungsweise als [FeIII(H2O)5(NO−)]2+ angesehen werden kann. Der Ligand ist in dieser Formulierung ein NO−-Ion, das im Triplett-Zustand vorliegt, also als Teilchen mit zwei ungepaarten Elektronen. (Können Sie diese Aussage in die bisherigen Erfahrungen einordnen?)
Versuch 23.1.30: Bildung von Aqua-nitrosyl-eisen(II)-Ionen aus (a) Nitrit und (b) Nitrat (Ringprobe)
• 3 HNO2 → 2 NO + HNO3 + H2O
• [Fe(H2O)6]2+ + NO → [Fe(H2O)5(NO)]2+ + H2O
• bei HNO3 zuerst: 2 Fe2+ + HNO3 + 2 H3O+ → 2 Fe3+ + HNO2 + 3 H2O
Phosphor gehört zu den Elementen, die Allotrope mit sehr unterschiedlichen Eigenschaften bilden.
Die reaktivste Form ist der weiße Phosphor, P4. Die tetraedrischen Moleküle folgen hinsichtlich ihrer Bindungssituation den schon behandelten Regeln. Als Element der dritten Periode beteiligt Phosphor praktisch nur die p-Orbitale an den Element-Element-Bindungen (6 % s-, 94 % p-Anteil). Da der 90°-Winkel zwischen den Orbitalen stark vom 60°-Winkel auf den Tetraederflächen abweicht, sind die P-P-Bindungen massiv gebogen („Bananenbindungen“, engl. banana bonds). Damit einher geht eine hohe Ringspannung und eine hohe Reaktivität. Das folgende Bild zeigt links das P4-Molekül, in der Mitte eine der sechs gebogenen p-p-Bindungen und rechts eines der vier freien Elektronenpaare mit nahezu reinem s-Charakter.
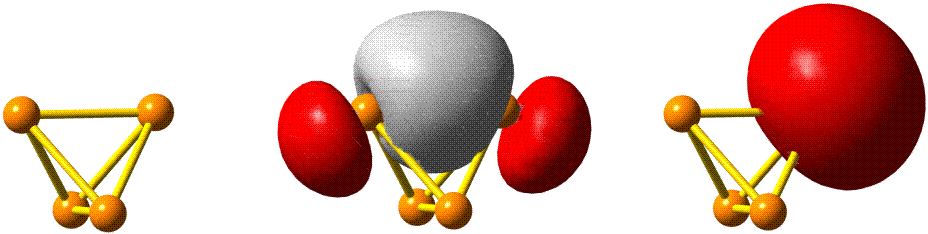
Beim Erhitzen von weißem Phosphor entsteht der erheblich weniger reaktive rote Phosphor, aus dem sich das geordnet kristallisierende Allotrop des violetten oder Hittorfschen Phosphors herstellen lässt.
Der Aufbau des Hittorfschen Phosphors ist recht kompliziert, indem zwei nicht-kovalent verbundene Teilstrukturen ineinander verschachtelt sind, deren jede aus Röhren besteht, die parallel und senkrecht zueinander angeordnet sind. Im Bild ist unten eine Röhre in Seitenansicht gezeigt; mit dieser verbunden sind die beiden senkrecht verlaufenden Röhren darüber rechts und links. Die obere mittlere Röhre gehört zur anderen Hälfte der Struktur. Ist die Gesamtstruktur komplex, so entspricht doch die Umgebung eines einzelnen Phosphoratoms der Erwartung: Bindungswinkel von ca. 100° spiegeln wie beim Schwefel Bindungen über p-Orbitale wider, die durch die Abstoßung der nächsten Nachbarn um einige Grad auseinander gedrängt sind.
Die stabile Modifikation ist der schwarze Phosphor. Während die mittleren Atomabstände und Bindungswinkel dem violetten Phosphor entsprechen, ist der Aufbau aus vernetzten P6-Ringen mit Sesselkonformation sehr viel geordneter.
Die Reaktivität des weißen Phosphors unterscheidet sich erheblich von derjenigen der anderen Allotrope. Vor allem die leichte Oxidierbarkeit erfordert die Beachtung besonderer Sicherheitsregeln beim Umgang mit weißem Phosphor.
Versuch 23.2.2: Brennbarkeit von weißem Phosphor
• P4 + 5 O2 → P4O10
Versuch 23.2.5 oder 6: Phosphoreszenz bei der Reaktion von Phosphordampf mit kleinen Mengen Sauerstoff.
Obwohl isotyp (dem gleichen Strukturtyp angehörend), werden die Strukturen von Arsen, Antimon und Bismut in vielen Lehrbüchern unterschiedlich dargestellt. Bei der Arsenstruktur wird in der Regel eine Stapelung gewellter Sechsringschichten betont. Im Vergleich zu den Sesseln der Diamant- oder Blendestruktur ist die starke Wellung auffallend – eine Folge des geringen s-Anteils der Bindungen (siehe unten). Die Formulierung isolierter Schichten ist nicht völlig berechtigt. Ober- und unterhalb eines jeden Sechsrings befindet sich jeweils ein Atom, dessen Abstand zu den Atomen des Ringes nur um ca. 24 % größer ist als die As-As-Bindungslänge (gestrichelte Linien in der Abbildung). Zusammen bilden die acht Atome einen verzerrten Würfel.
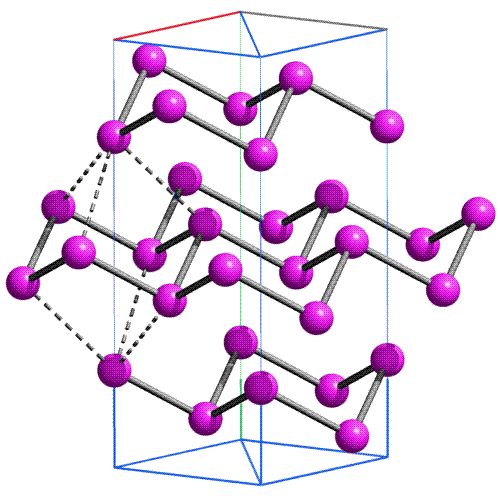
Dessen Verzerrung ist bei den schweren Homologen des Arsens kleiner (ca. 15 % Abstandsunterschied), so dass die Antimon- und die Bismutstruktur meist mit den zusätzlichen Bindungen dargestellt wird:
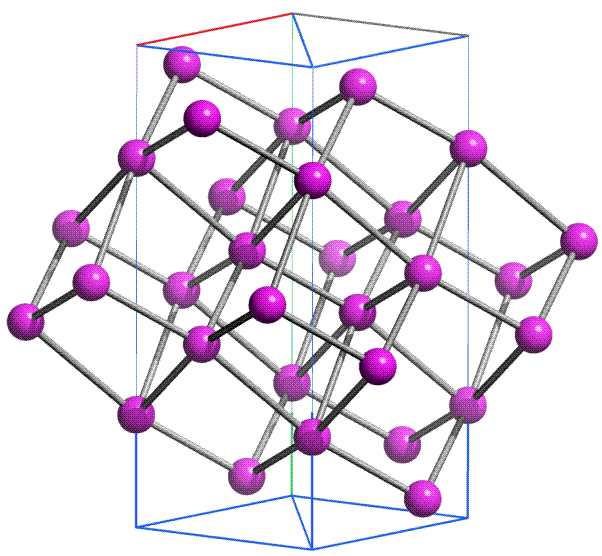
Wären in diesen Strukturen alle Bindungen gleich lang und alle Bindungswinkel 90°, läge die α-Polonium-Struktur vor, die kubisch primitive Kugelpackung. Die Abweichungen von der idealisierten Struktur sind in der folgenden Tabelle zusammengestellt.
| Pschwarz | As | Sb | Bi | |
|---|---|---|---|---|
| E-E-Bindung/pm | 223,0 | 251,7 | 290,8 | 307,1 |
| E···E-Kontakt/pm | – | 311,9 | 335,4 | 352,8 |
| Kontakt/Bindung | ca. 1,5 | 1,239 | 1,153 | 1,149 |
| E-E-E/° | 100,2 | 96,6 | 95,6 | 95,5 |
Die Eigenschaften der drei Elemente sind bemerkenswert. Ihre Sprödigkeit sowie die vor allem bei Bismut deutliche Dichteanomalie zeigen gerichtete Bindungen an, trotzdem weisen sie typische Metalleigenschaften auf, die wir mit Elektronenmangel verknüpft hatten: eine recht hohe elektrische Leitfähigkeit und den typischen Metallglanz.
Versuch 23.3.?: Sprödigkeit von Antimon
Versuch 23.3.?: Dichteanomalie bei Bismut
• flüssiges Bismut sprengt beim Erstarren ein Glasgefäß: die Dichte des Feststoffs ist wie beim Wasser am Erstarrungspunkt kleiner als die Dichte der Flüssigkeit.
Um die Bindungsverhältnisse im elementaren Arsen zu beschreiben, ist zuerst ein Blick auf eine einzelne Schicht sinnvoll. Jedes Arsenatom hat innerhalb der Schicht drei nächste Nachbarn. Der Bindungswinkel ist nur wenig größer als 90° – auch hier verursacht durch Abstoßung zwischen den Atomen. Eine Beteiligung von s-Orbitalen kann vernachlässigt werden. Eine isolierte Arsenschicht ist demnach zwar ein Riesenmolekül, es wird aber auf dieselbe Weise zusammengehalten wie ein S8-Ring oder ein Cl2-Molekül. Die einzelne Arsenschicht sollte daher keine metallischen Eigenschaften haben (keine elektrische Leitfähigkeit, kein Metallglanz), da ein metalltypischer Elektronenmangel nicht erkennbar ist – bei Cl2, S8 und der Arsenschicht ist vielmehr mit normalen 2e-2z-Bindungen die Oktettregel erfüllt.
Was ändert sich an diesem Bild, wenn nun Wechselwirkungen zwischen den Schichten berücksichtigt werden? Hier wiederholt sich auf dem Weg von Arsen zu Bismut eine Strukturänderung, die bereits bei in der 16. Gruppe auffällig war: Alle Atomabstände werden größer, der Unterschied zwischen den Bindungen und den übernächsten Atomkontakten wird jedoch – wie gezeigt – kleiner. Werden mit der Annäherung der Atomschichten auch die Eigenschaften metallischer, das heißt, gewinnen die Elektronen an Beweglichkeit?
Um dies zu untersuchen, betrachte man ein einzelnes Arsen-, Antimon- oder Bismutatom in der Strukturabbildung mit seinen drei nächsten und den drei übernächsten Nachbarn, erstere in derselben Schicht, letztere aus der benachbarten. Die aufeinander praktisch senkrecht stehenden Bindungen zu den Nachbaratomen derselben Atomschicht definieren die Lage der drei p-Orbitale des herausgegriffenen Atoms. Die drei Atome der Nachbarschicht liegen nun recht genau in der Verlängerung eines jeden p-Orbitals nach „hinten“, also auf der Gegenseite der Bindung. Auf den gesamten Kristall bezogen bedeutet dies, das jedes px-Orbital Teil einer unendlichen linearen Kette aus px-Orbitalen ist, das Gleiche gilt für py- und pz-Orbitale.
Im folgende Schema ist für eines der p-Orbitale des Arsen die Folge dieser Wechselwirkung von einem einzelnen Atom ausgehend (links) bis hin zur unendlich ausgedehnten Kette entwickelt. Rechts neben dem einzelnen Atom ist eine isolierte As-As-Bindung gezeichnet (besetztes σ-MO, leeres σ*-MO). Weiter rechts sind die As-As-Bindungen Teil einer Kette mit längeren Kontakten zwischen den As-Atomen (gepunktet) als in den Bindungen (Striche). Sind Bindungen und übernächste Kontakte gleich lang, so resultiert eine mittlere Bindungsordnung von ½ in der dann äquidistanten Kette. Die sich ausbildenden Orbitale und ihre Energien sind schematisch angedeutet: aus n einfach besetzten Atomorbitalen in der Kette entsteht ein Band aus n/2 bindenden und n/2 antibindenden MOs; n/2 Elektronenpaare besetzen die bindenden MOs, während die weniger stabile obere Bandhälfte frei bleibt.
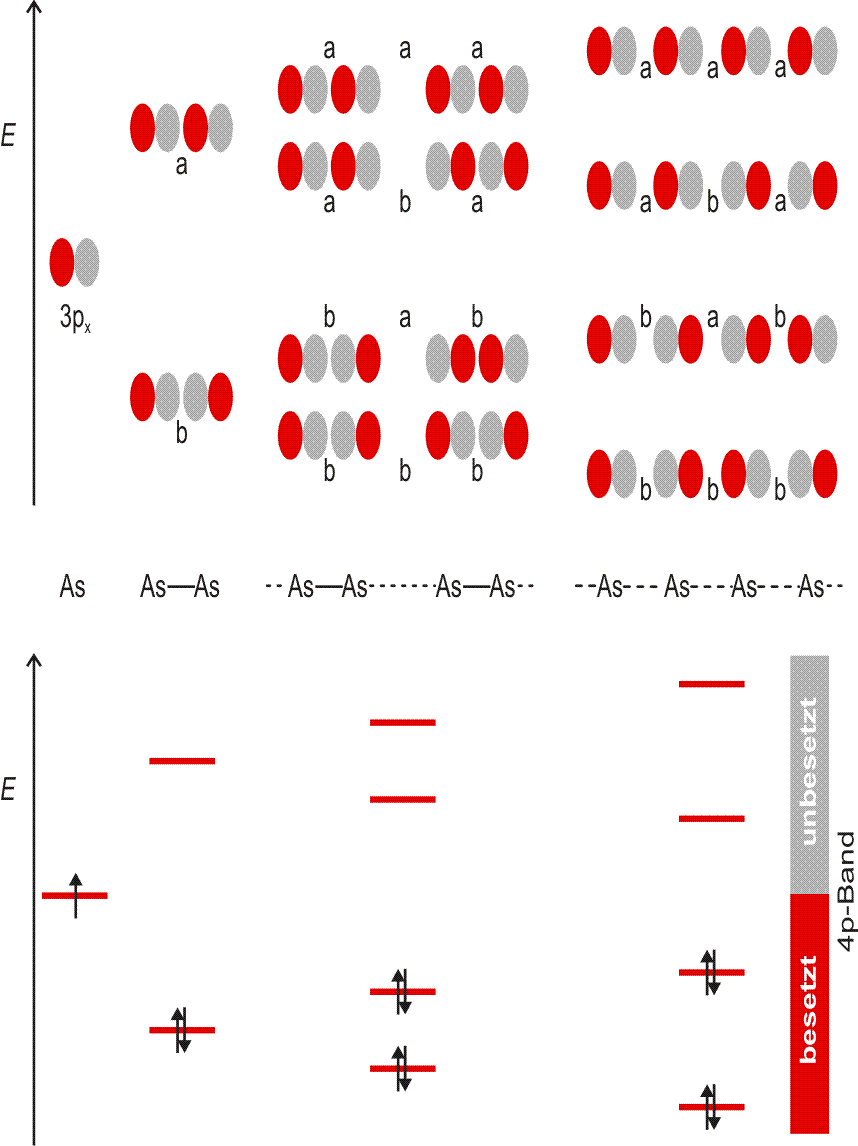
Das hier in einer Dimension entwickelte Bändermodell zeigt für Arsen metallische Eigenschaften an: zahlreiche elektronische Anregungen aus dem besetzten in den leeren Teil des Energiebandes mit allen möglichen Energiewerten erklären die Absorption aller Energien (Farben) des sichtbaren Lichts – Arsen glänzt metallisch. Auch sehr kleine Energien führen zur Anregung von Elektronen in einen leitenden Zustand – die elektrische Leitfähigkeit des Arsens liegt im für Metalle charakteristischen Bereich.
Da sich auf der anderen Seite bei den nicht so elektronenarmen Elementen der 15. Gruppe aus dem Orbitalbild gerichtete Bindungen ergeben, kann Duktilität nicht erwartet werden.
Das Ergebnis der Betrachtung: Arsen, Antimon und Bismut zeigen in ihren Strukturen Koordination von Nachbaratomen über die kovalente Bindigkeit hinaus – und damit Anfänge eines Elektronengases, indem Richtungen metalltypischer Elektronendelokalisation entstehen.
Die große Neigung des Phosphors zur Koordinationszahl vier wird deutlich, wenn die Homologen der Verbindungen mit N+III und N+V, salpetrige Säure und Salpetersäure, betrachtet werden: „phosphorige Säure“ und Phosphorsäure, außerdem die „hypophosphorige Säure“, zu der es in der Stickstoffchemie kein beständiges Homolog gibt.
Versuch 23.2.11: Disproportionierung von weißem Phosphor in Lauge
• 4 P4 + 12 OH− + 12 H2O → 4 PH3 + 12 H2PO2−
Versuch 23.2.10: Disproportionierung von H3PO3
• 4 H3PO3 → PH3 + 3 H3PO4
Bei den verschiedenen Versuchen war das als PH3 formulierte Gas einmal selbstentzündlich, ein anderes Mal nicht. Die Ursache ist, dass reines, nicht selbstentzündliches PH3 nur unter besonders kontrollierten Bedingungen entsteht (zum Beispiel bei der Protolyse von Phosphoniumsalzen bei niedriger Temperatur, im Alltag bei der Reaktion des bei der Wühlmausbekämpfung eingesetzten Aluminiumphosphids, AlP, mit Wasser). Ansonsten treten weitere Phosphane im Gemisch mit PH3 auf, vor allem das dem Hydrazin entsprechende Diphosphan, P2H4. Wie alle anderen höheren Phosphane geht die leichte Entzündlichkeit mit dem Vorliegen von P-P-Bindungen einher – man vergleiche die Eigenschaften des weißen Phosphors. Höhere Phosphane sind in großer Zahl bekannt, ähnlich wie sich zwischen Kohlenstoff und Wasserstoff zahreiche Alkane formulieren lassen. Bei einem Vergleich der Strukturen von Alkanen und Phosphanen fällt auf, dass die Phosphorverbindungen wie schon das Element selbst zur Bildung kleiner Ringe neigen (man vergleiche den kleineren Bindungswinkel in Phosphanen mit dem Tetraederwinkel in Alkanen). Ein typisches Strukturmotiv liegt zum Beispiel im P7H3 vor:
Wasserstoffverbindungen treten auch bei den schweren Homologen des Phosphors auf. Besondere Bedeutung hat Arsan, AsH3. Es entsteht unter den Bedingungen der Marshschen Probe, die von besonderer Bedeutung in der Geschichte der Gerichtsmedizin ist.
Versuch 23.3.?: Herstellung und Zersetzung von Arsan, AsH3
• As2O3 + 6 Zn + 12 H3O+ → 2 AsH3 + 6 Zn2+ + 15 H2O
Bei den Versuchen zur Disproportionierung von weißem Phosphor und von „phosphoriger Säure“ entstehen neben Monophosphan weitere Produkte, in denen P-H-Bindungen enthalten sind. So zeigt die Strukturanalyse, dass der Aufbau von H3PO3 nicht mit dem Namen „phosphorige Säure“, also P(OH)3, übereinstimmt, sondern dass vielmehr ein Tautomer der phosphorigen Säure vorliegt. Unter Tautomerie wird eine besondere Form der Isomerie verstanden, bei der sich die verschiedenen Isomere nur durch unterschiedliche Positionen von Wasserstoffatomen unterscheiden. So liegt eines der drei Wasserstoffatome in der kristallinen Verbindung nicht an einem Sauerstoffatom, sondern am Phosphoratom gebunden vor; diese Form heißt Phosphonsäure:
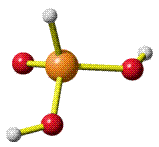
Die zugehörigen Lewisformeln sind links (das erwartete Tautomer – in Klammern, da es im Lösungsgleichgewicht nicht nachweisbar ist) und rechts dargestellt:
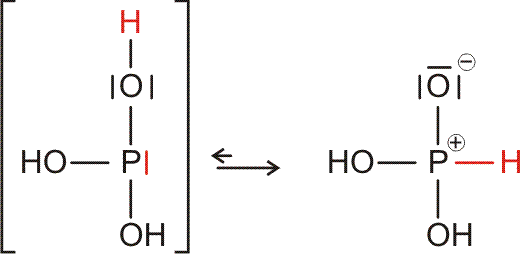
Der unerwartete Aufbau der kristallinen Phosphonsäure führt natürlich zu der Frage, ob solche Tautomere auch in der verwandten Schwefelchemie möglich sind. Ein Kandidat ist H2SO3. Hier ist die Isolation der reinen Säure bisher noch nicht gelungen, es konnte jedoch gezeigt werden, dass H2SO3 im Lösungsgleichgewicht in beiden tautomeren Formen vorkommt, der schwefligen Säure (links) und der Sulfonsäure (rechts):
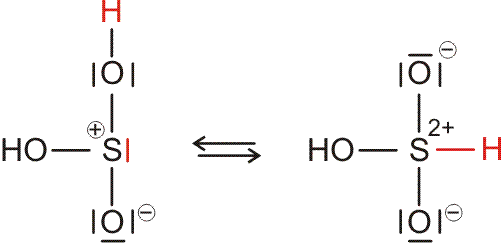
Isoliert wurden Salze mit dem HSO3−-Ion und tatsächlich werden hier sowohl Sulfite als auch Sulfonate gefunden. Die Salze der Phosphonsäure liegen dagegen stets in der Form der Phosphonate und nicht als Phosphite vor, H3PO3 ist daher eine zweibasige Säure.
Die Neigung des Phosphors zur Koordinationszahl vier wird besonders deutlich, wenn das bei der Umsetzung von weißem Phosphor in starker Lauge entstandene H2PO2−-Ion strukturell untersucht wird. Hier der Aufbau des Ions in der Kristallstruktur des Bariumsalzes:
Die freie Säure ist damit eine reduzierte Form der Phosphonsäure und zwar Phosphinsäure:
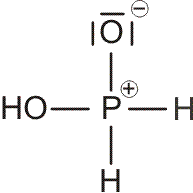
Phosphonsäure entspricht mit der Oxidationstufe +III für Phosphor der salpetrigen Säure. Der strukturelle Unterschied – die Koordinationszahl drei bei der Stickstoffverbindung, aber vier bei Phosphonsäure, außerdem das Fehlen von N-H-Bindungen – ist typisch und bestimmt die Unterschiede bei dem in der Praxis wichtigsten Paar: Salpetersäure und Phosphorsäure.
Versuch 23.2.12: P4O10 als Säureanhydrid
Nichtmetalloxide sind Säureanhydride. Mit Wasser reagieren sie unter Bildung der Säuren mit dem jeweiligen Element in derselben Oxidationsstufe. Bei der Reaktion mit Wasser findet eine der beiden folgenden Reaktionen statt:
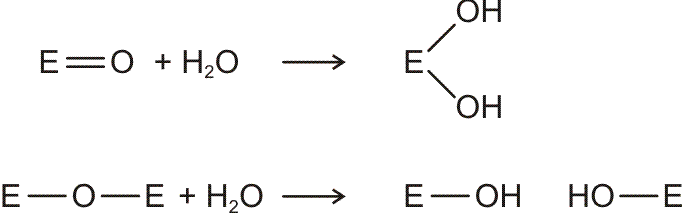
Beispiele sind die Reaktionen beim Auflösen von SO2 oder SO3 in Wasser. In der Stickstoffchemie führt die Umsetzung der wenig stabilen Oxide N2O3 und N2O5 mit Wasser zu salpetriger Säure und Salpetersäure.
Das stabilste Phosphoroxid ist P2O5, das in mehreren Modifikationen auftritt. neben einer polymeren Form existiert ein P4O10, dessen P4O6-Gerüst Adamantanstruktur (C10H16) hat, die auch in der Diamant- und Zinkblendestruktur ausgebildet wird.
Durch nukleophilen Angriff von Wassermolekülen auf die Phophorzentren bildet sich Phosphorsäure (auch: Orthophosphorsäure; „Ortho…“ bezeichnet die wasserreichste Form).
P4O10 + 6 H2O → 4 H3PO4
Diese bei Raumtemperatur stark exotherm ablaufende Reaktion ist bei erhöhter Temperatur teilweise umkehrbar. Bei Phosphor von besonderer Bedeutung ist die Wasserabspaltung zwischen zwei Phosphorsäuremolekülen, die intermolekulare Kondensation, die als nukleophiler Angriff eines phosphorgebundenen Sauerstoffatoms auf ein benachbartes Phosphoratom abläuft:
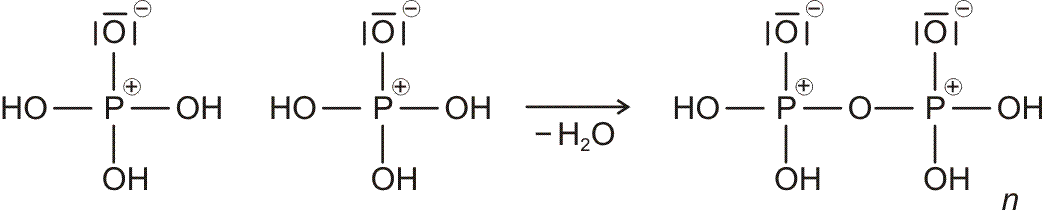
Bei Temperaturen von ca. 200 °C führt die Polykondensation zu einem linearen Polymer der Summenformel HPO3, der Metaphosphorsäure:
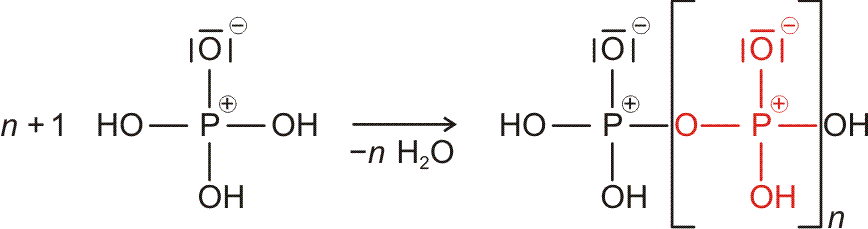
Die Summenformel der Metaphosphorsäure zeigt dasselbe Atomzahlverhältnis wie bei der Salpetersäure. Der Strukturunterschied ist dabei typisch: mit dem kleineren Stickstoffatom ist die Koordinationszahl drei realisiert, es treten N-O-Doppelbindungen auf. Die Doppelbindungen werden von p-Orbitalen aufgebaut („pπ-pπ-Bindungen“). Das größere Phosphoratom hat dagegen vier Bindungspartner, Doppelbindungen werden vermieden. Diese Regeln gelten auch bei anderen Elementen der dritten Periode, wobei die Doppelbindungsregel, nach der diese Elemente pπ-pπ-Bindungen vermeiden, zahlreiche Ausnahmen kennt, zum Beispiel bei SO2 und SO3.
Die Kondensation von Phosphorsäure und die Spaltung von Polyphosphat ist eine bedeutende Reaktion sowohl in der belebten Natur (ATP als universeller Energiespeicher) als auch in der Technik. Ein großtechnisches Produkt ist zum Beispiel Na5P3O10, das Natriumtriphosphat. Es wird als Zusatz für technische Reinigungsmittel hergestellt. Triphosphat ist ein guter Ligand für Calciumionen, die für die Wasserhärte verantwortlich sind. Zur technischen Herstellung wird Natronlauge und Phosphorsäure im passenden stöchiometrischen Verhältnis gemischt und zur Kondensation auf ca. 150 °C erhitzt:
3 H3PO4 + 5 NaOH → Na5P3O10 + 7 H2O
Das P3O105−-Ion hat in der Kristallstruktur des Natriumsalzes den folgenden Aufbau:
Versuch 23.2.17: Triphosphat als Ligand für Calcium
Polykondensation ist nicht darauf beschränkt, Isopolysäuren, das sind kondensierte Polysäuren, die aus einem einzigen Grundbaustein bestehen, zu bilden. Heteropolysäuren und deren Salze bilden sich, wenn in einer Lösung verschiedene zur Kondensation neigende Säuren miteinander reagieren. Ein Beispiel ist die Polykondensation von Molybdänsäure (≈ H4MoO5) und Phosphorsäure. Die Bildung des Ammoniumsalzes des [P(Mo3O10)4]3−-Ions wird zum Phosphatnachweis genutzt.
Im abgebildeten Anion ist formal jedes O-Atom eines Phosphat(3−)-Ions durch ein Mo3O10-Fragment ersetzt. Diese formale Betrachtung führt allerdings nicht sehr weit, da die dreikernigen Molybdäneinheiten nicht als individuelle Baugruppen erkennbar sind, sondern als dreidimensional vernetztes Aggregat das Phosphoratom regelrecht einkapseln.
Versuch 23.2.16: Bildung von Molybdophosphat (Phosphomolybdat, Phosphododekamolybdat)
Stickstoff in Form von Ammoniumsalzen, Harnstoff und Nitrat sowie Kalium und Phosphat sind die wesentlichen Bestandteile von Düngemitteln. Als Rohstoff für Phosphatdünger dienen die in großen Lagern vorkommenden Calciumphosphate, die zum Teil aus „biologischem Apatit“ entstanden sind, der mineralischen Komponente von Knochen.
Bei der Nutzung von Düngemitteln auf Calciumphosphatbasis ist die Löslichkeit der zugänglichen Verbindungen in Wasser entscheidend:
• Calcium-dihydrogenphosphat („primäres Calciumphosphat“), Ca(H2PO4)2, wird aufgrund seiner guten Löslichkeit in Wasser als schnell aufnehmbarer Dünger verwendet,
• Calciumhydrogenphosphat („sekundäres Calciumphosphat“), CaHPO4, das ebenfalls als löslicher Dünger verwendet werden kann, und
• Calciumphosphat („tertiäres Calciumphosphat“), Ca3(PO4)2, von dem sich in Wasser schwerlösliche Phosphatminerale (Phosphorit, Apatit) herleiten.
Der wichtigste Rohstoff für die technische Herstellung von Phosphorverbindungen ist Fluorapatit, Ca5(PO4)3F („3 Ca3(PO4)2·CaF2“), in dem die Phosphatkomponente von Calciumfluorid begleitet wird.
Aufschlussphosphorsäure wird durch Umsetzung von Apatit mit Schwefelsäure gewonnen. Mit dem Calciumphosphatanteil des Apatits formuliert ergibt sich als Reaktionsgleichung:
Ca3(PO4)2 + 3 H2SO4 → 3 CaSO4 + 2 H3PO4
Die entstehende Phosphorsäure wird für die weitere Verwendung gereinigt und wird dann vor allem für die Herstellung des Calciumphosphatdüngers „Tripelsuperphosphat“ eingesetzt.
Thermische Phosphorsäure wird hergestellt, indem im ersten Schritt bei 1400–1500 °C Apatit mit Kohle und Kies umgesetzt wird. Phosphat wird unter diesen Bedingungen durch Kohlenstoff zu Phosphor reduziert, während das verbleibende Calciumoxid (Ca3(PO4)2 = „3 CaO·P2O5“ durch SiO2 verschlackt wird):
Ca3(PO4)2 + 3 SiO2 + 5 C → 3 CaSiO3 + 2 P + 5 CO
Phosphor fällt bei diesem Verfahren als weißer Phosphor an. Ca. 85 % des so gewonnenen weißen Phosphors werden verbrannt und das entstehende Phosphorpentoxid wird mit Wasser zu Phosphorsäure umgesetzt. Die entstehende Phosphorsäure ist recht rein und wird zum Beispiel auch für die Waschmittelproduktion und für die Lebensmitteltechnik eingesetzt.
Der restliche Phosphor wird zu weiteren Phosphorverbindungen umgesetzt (Phosphorpentasulfid, P2S5, und Phosphorsulfidchlorid, PSCl3, für die Herstellung von Pflanzenschutzmitteln; Phosphortrichlorid, PCl3, Phosphoroxichlorid, POCl3, und Phosphorpentachlorid, PCl5 (siehe weiter unten), als Grundchemikalien für die organische Chemie).
Das in Wasser leicht lösliche Ca(H2PO4)2 wird nach dem gleichen Prinzip hergestellt wie Aufschlussphosphorsäure, nämlich durch Protonierung der Base Phosphat(3−):
Superphosphat entsteht bei der Umsetzung von Apatit mit einer geringeren Menge Schwefelsäure, als bei der Herstellung von Aufschlussphosphorsäure verwendet wird (1 statt 1,5 mol Schwefelsäure pro mol Phosphat):
Ca3(PO4)2 + 2 H2SO4 → 2 CaSO4 + Ca(H2PO4)2
Superphosphat ist ein Dünger von beschränktem Wert, da die Beimengung von Gips einen unwirtschaftlich niedrigen Phosphatgehalt bedingt; darüber hinaus ist die Anreicherung landwirtschaftlich genutzter Böden mit Gips (Calciumsulfat-Dihydrat) nicht unproblematisch. Tripelsuperphosphat ist dagegen frei von größeren Mengen nutzloser Beimengungen, da hier als Säure Phosphorsäure, meist Aufschlussphosphorsäure, verwendet wird. Das gewünschte Dihydrogenphosphat entsteht hier sowohl durch die Protonierung von Phosphat als auch durch die Deprotonierung der Säure:
Ca3(PO4)2 + 4 H3PO4 → 3 Ca(H2PO4)2
Beim elektronegativen Element Stickstoff ist schon die übliche Bezeichnung irreführend. Nur die N-F-Bindung ist zum Halogen hin polarisiert, so dass Fluor die Oxidationszahl -I erhält. N-Cl-Bindungen sind dagegen weitgehend unpolar, während NBr3 ein Bromnitrid darstellt. Hierdurch scheint NBr3 Ähnlichkeit zu NH3 zu bekommen, doch die Reaktivität der Bromverbindung ist völlig verschieden von derjenigen der Base Ammoniak. Das folgende Bild zeigt die Besonderheiten des NBr3.
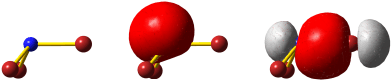
NBr3 (links), das freie Elektronenpaar am Stickstoffatom mit 70 % s-Anteil (Mitte), eine der drei N-Br-Bindungen mit 10 % s-Anteil bei Stickstoff Bindungswinkel: 108,4° (Techn. Anm.: b3lyp/6-311+g(2d,p) + nbo)
Die Rechnung bestätigt die negative Beladung des N-Atoms, das je nach Rechenmethode Ladungen von −0,5 bis −0,7 erhält. Im Unterschied zu Ammoniak hat die N-Br-Bindung weitgehenden p-Charakter, so dass der nahe beim Tetraederwinkel liegende Bindungswinkel nicht mit einem Ammoniak-typischen s-Anteil einhergeht. Der dadurch hohe s-Anteil des freien Elektronenpaars am Stickstoffatom lässt keine nennenswerte Basizität zu. Der große Bindungswinkel kommt durch die Abstoßung der großen Bromatome zustande – das Molekül ist gespannt und äußerst reaktiv. Brom- und vor allem Iodnitrid sind daher explosive Verbindungen (beachten Sie die einfache Bildung aus laborüblichen Reagenzien und die damit verbundene Unfallgefahr!):
Versuch 23.1.33: Herstellung und Eigenschaften von Iodstickstoff-Ammoniak-Addukten
• 5 NH3 + 3 I2 → NI3·NH3 + 3 NH4I
Die drei bekannten Phosphorpentahalogenide weisen eine besonders abwechslungsreiche Strukturchemie auf. Während mit dem großen Brom-Substituenten ein PBr4+Br− gebildet wird, hängt der Aufbau von Phosphorpentachlorid vom Aggregatzustand ab. Das durch Reaktion von PCl3 mit Chlor als Feststoff entstehende PCl5 liegt als PCl4+PCl6− vor, es bildet jedoch im Gaszustand und in Lösung PCl5-Moleküle. Als trigonal bipyramidales Molekül liegt auch PF5 vor. Die unterschiedlichen Bindungslängen werden verständlich, wenn entlang der beiden axialen Substituenten eine 4e-3z-Bindung mit geringerer kovalenter Bindungsordnung formuliert wird als zwischen den äquatorialen P-F-Bindungen.
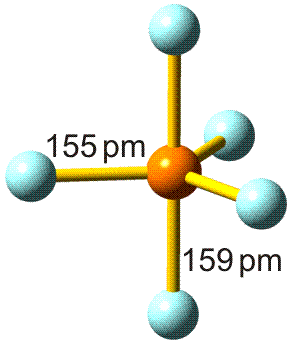
Versuch 23.2.19a: Bildung von Phosphor(V)-chlorid aus Phosphor(III)-chlorid und Chlor
• PCl3 + Cl2 → PCl5
Auch bei den Halogeniden ist der Übergang vom elektronegativen Nichtmetall Stickstoff zum elektropositiven Metall Bismut sichtbar. Zwischen der explosiven Molekülverbindung NCl3 und den typischen Säurechloriden PCl3 und AsCl3 (beide Verbindungen hydrolysieren in Wasser zu H3PO3 beziehungsweise H3AsO3) auf der einen Seite und dem schon fast salzartigen BiCl3 zeigt das Antimontrichlorid mittlere Eigenschaften: es hydrolysiert beim Lösen in Wasser und das entstehende Sb(OH)3 ist amphoter.
Versuch 23.3.?a: Eigenschaften von SbCl3
Der Metall-Nichtmetall-Übergang verläuft in der 14. Gruppe besonders spektakulär. Die Diamantstruktur führt zu der Besonderheit, dass sich ein Valenzband und ein Leitungsband trennen – Silicium und Germanium sind Halbleiter. – Wem die Strukturchemie eines einatomigen Anions wie Chlorid zu fad war: die 14. Gruppe hält Silicate bereit: Inselsilicate, Kettensilicate, Bandsilicate, Schichtsilicate, Alumosilicate, …
Wie Phosphor, so zeigt auch Kohlenstoff eine besonders eindrucksvolle Polymorphie: der elektrische Leiter Graphit als stabiles Allotrop, der nur unwesentlich unstabilere Nichtleiter Diamant und die extrem unstabilen, aber hinsichtlich der Umwandlung in Graphit inerten Fullerene.
Graphit besteht aus ebenen Schichten, in denen die Kohlenstoffatome in einer Umgebung vorliegen, die den polycyclischen Aromaten gleicht (oder umgekehrt: die polycyclischen Aromaten sind Ausschnitte aus einer Graphitschicht). Elektrische Leitfähigkeit liegt innerhalb der Schichten vor, nicht zwischen diesen. Da der elektronisch gesättigte σ-Anteil des Wabengerüsts einer Schicht – hier liegen 2e-2z-Bindungen vor – keine Orbitalüberlappung mit den senkrecht stehenden p-Orbitalen aufweist (die Bindungssysteme sind orthogonal), betrachten wir nur die letzteren als Träger der Leitfähigkeit. Eine Verständnis für die zweidimensionale Leitfähigkeit entsteht, wenn das jeweils eine p-Orbital pro Kohlenstoffatom als Teil eines halbgefüllten p-Bandes betrachtet wird.
Beim Verdampfen von Graphit entstehen Fullerene und Kohlenstoff-Nanoröhren. Die Bindungsprinzipien beider Substanzklassen modifizieren die elektronischen Verhältnisse in Graphit. Im folgenden Bild ist das besonders hochsymmetrische [60]Fulleren, C60 dargestellt.
(Hier erst mal nur ein Test, wenn es klappt, sieht man ein bewegtes Molekülmodell.)
In der Kristallstruktur nähern sich die einzelnen C60-Moleküle ihren Nachbarn auf ca. 330–340 pm, was auch bei den intermolekularen Kräften die Ähnlichkeit zu Graphit hervorhebt. Könnten Sie übrigens ein sinnvolles Packungsprinzip für C60-Moleküle vorhersagen? (Hier die Auflösung.)
Kohlenstoff-Nanoröhren stellen eine besonders intensiv untersuchte Gruppe von Kohlenstoff-Allotropen dar. Ihr Aufbau zeigt mehr Verwandtschaft mit Graphit. Sie entstehen formal (möglicherweise auch in der Realität), indem sich einzelne Graphitschichten aufrollen und an den Nahtstellen verschmelzen.
Diamant, dessen Tetraederstruktur bei Kohlenstoff, Silicium und Germanium und α-Zinn („graues Zinn“) auftritt, ist ein typischer Nichtleiter. Die extrem hohe und technisch genutzte Wärmeleitfähigkeit des Diamant zeigt, dass diese Eigenschaft für Metalle typisch ist, dass aber nicht gilt, dass ein Stoff ein Metall ist, wenn hohe Wäremeleitfähigkeit auftritt.
Nach der 8−N-Regel ist als niedrigste Oxidationsstufe des Kohlenstoffs −IV möglich. Diese wird in Aluminiumcarbid erreicht, das wie das großtechnisch hergestellte Calciumcarbid zu den salzartigen Carbiden gerechnet wird. Bei der Umsetzung mit Säure, Wasser oder Alkalilauge wird die außerordentlich starke Base C4− durch Wasser zu Methan protoniert.
Versuch 24.1.4: a) Methan aus Aluminiumcarbid und Lauge
• Al4C3 + 12 H2O + 4 OH− → 4 [Al(OH)4]− + 3 CH4
Versuch 24.1.6: a) Prinzip der Grubenlampe. b) „Schlagende Wetter“
Methan wird als „Erdgas“ gefördert. Es ist der einfachste Kohlenwasserstoff und zeigt das auch für den Aufbau des Diamants wichtige Prinzip, dass zur Erfüllung der Oktettregel auch die s-Elektronen in vollem Umfang an den Bindungen teilnehmen. Beachten Sie, dass der in Lehrbüchern bisweilen erwähnten „Promotionsenergie“, der Energie zur Anregung des 2s22p2-Grundzustands des Kohlenstoffatoms zum 2s12p3-Zustand, die Spinpaarungsenergie gegenübersteht, welche den Zustand mit vier ungepaarten Elektronen stabilisiert.
Dass es sinnvoll ist, allein mit der Oktettregel zu argumentieren, wird auch durch Methylen, CH2 („Carben“), verdeutlicht. Methylen ist so unstabil, dass es nur als Zwischenstufe bei chemischen Reaktionen formuliert wird. Das reaktive Molekül liegt dabei keineswegs im erwarteten Singulett-Zustand vor, sondern es ist ein Triplett-Molekül, dessen Orbitale weitgehend denjenigen des Wassermoleküls entsprechen, mit dem Unterschied, dass die beiden spintragenden Orbitale im Wasser doppelt besetzt sind (die beiden freien Elektronenpaare am Sauerstoffatom).
Die Formel des durch Reaktion von Kohle mit Branntkalk (siehe unten) gewonnenen Calciumcarbids passt nicht zu einer Verbindung aus Ca2+ und C4−-Ionen. Der salzartige Feststoff besteht vielmehr aus Calcium- und Acetylid-Ionen. (Im folgenden Bild ist nicht die tetragonal-raumzentrierte Elementarzelle dargestellt, sondern eine doppelt so große Zelle, die den Bezug zur Natriumchloridstruktur hervorhebt.)
Bei der Reaktion mit Wasser entsteht Ethin (Acetylen), C2H2.
Versuch 24.1.4b: Ethin (Acetylen) aus Calciumcarbid und Wasser
• CaC2 + 2 H2O → C2H2 + Ca(OH)2
Die beiden technisch wichtigen Gleichgewichte erschließen sich aus ihren thermodynamischen Parametern. Ermitteln Sie, ausgehend von den Standardbildungsenthalpien, die Möglichkeiten, die Gleichgewichte zu verschieben (ΔfH°/kJ mol−1: CO −110, CO2 −393, H2Og −242).
Versuch 24.1.14: Reaktion von Wasserdampf und Kohle bei 900 °C
• C + H2O ⇌ CO + H2
Versuch 24.1.15: Reaktion von Kohlendioxid und Kohle bei 900 °C
• C + CO2 ⇌ 2 CO
Versuch 24.1.9a: Dichte und erstickende Wirkung von Kohlendioxid
Kohlendioxid ist das Anhydrid der Kohlensäure, H2CO3. Wie bei Salpetersäure wird die Lewis-Formel mit einer pπ-pπ-Doppelbindung formuliert; auch die Koordinationszahl drei des Zentralatoms ist die gleiche:
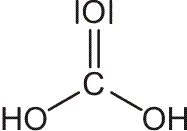
Kohlenmonoxid, ein nahezu unpolares Molekül, reagiert dagegen nicht mit Wasser. Es wird daher üblicherweise nicht als Anhydrid von H2CO2, der Ameisensäure, verstanden. Unter drastischeren Reaktionsbedingungen gelingt jedoch sowohl die Bildung von Formiat aus CO (Reaktion mit starker Lauge) als auch die Entwässerung der Ameisensäure:
Versuch 24.1.16: CO-Herstellung aus Ameisensäure und Schwefelsäure
• HCOOH → CO + H2O
Die Lewis-Formel von CO zeigt die Bedeutung der Oktettregel besonders eindringlich. Die rechte Grenzformel ist ohne Bedeutung.
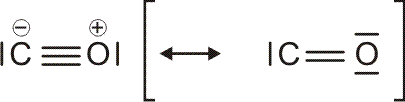
Wenn sich viele Atome ein-, zwei- oder dreidimensional zu einer ausgedehnten Struktur verbinden, führt die Überlappung der Atomorbitale zu Energiebändern. Sind die Bänder nur teilweise gefüllt oder kommt es durch die Überlappung von Bändern dazu, dass zwischen dem unstabilsten besetzten Zustand (in der Molekülchemie: HOMO von highest occupied molecular orbital) und dem stabilsten unbesetzten Zustand (in der Molekülchemie: LUMO von lowest unoccupied molecular orbital; in der Festkörperphysik liegt zwischen HOMO und LUMO das „Fermi-Niveau“) praktisch keine Energielücke auftritt, so liegt metallische Leitfähigkeit vor. Diese ist experimentell vor allem dadurch charakterisiert, dass sie mit der Temperatur abnimmt.
Liegt zwischen dem energiereichsten besetzten Zustand und dem energieärmsten unbesetzten Zustand eine größere Energielücke, so zeigt der betreffende Stoff das typische Nichtmetallverhalten: keine elektrische Leitfähigkeit, kein Metallglanz. Eine Energielücke bedeutet, dass das höchste besetzte Orbital das stabilere Band abschließt und dass alle Zustände des nächsthöheren Bandes unbesetzt sind. Besetztes und leeres Band heißen Valenzband und Leitungsband. Ist die Energielücke nur so groß, dass Elektronen durch thermische Energie oder durch Licht vom Valenz- in das Leitungsband angeregt werden können, liegt ein Halbleiter vor. Dieser ist experimentell vor allem dadurch gekennzeichet, dass seine (geringe) Leitfähigkeit mit der Temperatur zunimmt. Halbleitende Eigenschaften treten auf, wenn die Energielücke zwischen 0 und ca. 2 eV liegt. Diamant weist mit ca. 5 eV eine erheblich größere Lücke auf und ist daher kein Halbleiter. Das isotyp kristallisierende Silicium zeigt dagegen eine Bandlücke von ca. 1 eV. Da alle Wellenlängen des sichtbaren Lichts eine größere Energie tragen, werden alle Wellenlängen von Silicium absorbiert und regen dabei Elektronen vom Valenz- in das Leitungsband an. Der Halbleiter Silicium zeigt Metallglanz und seine elektrische Leitfähigkeit erhöht sich beim Bestrahlen.
Versuch 24.2.1b: Solarzelle
Die Ursache der Bandlücke bei den Elementen der 14. Gruppe diskutieren wir mit Hilfe eines Orbitalenergieschemas.
Die Reinheitsanforderungen an Halbleitersilicium sind extrem, da Verunreinigungen bereits in kleinster Menge die halbleitenden Eigenschaften des Siliciums stark verändern (Reinst-Si: Spez. Widerstand = 150.000 W cm; Si + 1 ppb Phosphor: 100 W cm; hierbei ist 1 ppb = „1 part per billion“ = 1 Teil auf 1 Milliarde, also zum Beispiel 1 mg auf 1000 kg). Die mehrstufigen technischen Prozesse zur Synthese von Reinstsilicium sind daher ungewöhnlich aufwendig.
Versuch 24.2.1a: Silicium aus Seesand und Magnesium
• 2 Mg + SiO2 → Si + 2 MgO
In der Technik wird anstelle des Magnesiums Kohlenstoff verwendet.
SiO2 + 2 C → Si + 2 CO
Ein Überschuss an Kohlenstoff wird vermieden, da sich dann vermehrt Siliciumcarbid, SiC, bilden würde. Das Rohsilicium wird über die Stufe des Trichlorsilans gereinigt, indem bei ca. 300 °C mit HCl-Gas umgesetzt wird:
Si + 3 HCl → SiHCl3 + H2
Anschließend wird die farblose Flüssigkeit durch Destillation gereinigt und bei ca. 1000 °C durch die Umsetzung mit Wasserstoff unter Umkehrung der Bildungsgleichung wieder zersetzt:
SiHCl3 + H2 → Si + 3 HCl
Das so erhaltene Silicium genügt in polykristalliner Form den Anforderungen an „Solarsilicium“.
Einkristalle aus hochreinem Halbleitersilicium werden durch Zonenschmelzen oder durch das Czochralski-Verfahren gewonnen (Kristallzucht aus einer Schmelze an einem „Impfkristall“).
Silicide entstehen bei der Umsetzung von Metallen mit Silicium. Mit starken Reduktionsmitteln wie Magnesium gelingt auch die Herstellung aus SiO2 und überschüssigem Metall:
Versuch 24.2.3: Magnesiumsilicid aus Seesand und Magnesium
• 4 Mg + SiO2 → Mg2Si + 2 MgO
Magnesiumsilicid kristallisiert in der „anti-Fluorit“-Struktur. Die Vorsilbe „anti“ weist darauf hin, dass gegenüber dem Prototyp, CaF2, Anion- und Kationplätze vertauscht sind.
Kristallstruktur von Mg2Si. Farbcode: graugrün Si, gelb Mg.
Die Hydrolyse von Siliciden unedler Metalle führt zu Silanen:
Versuch 24.2.4: Silan durch Hydrolyse von Magnesiumsilicid
• Mg2Si + 4 H2O → SiH4 + Mg(OH)2
Wie die Phosphorchloride ist auch Siliciumtetrachlorid kein Salz, sondern ein Säurechlorid.
Versuch 24.2.5: Hydrolyse von SiCl4
• SiCl4 + 4 H2O → H4SiO4 + 4 HCl
Kieselsäure, H4SiO4, genauer Orthokieselsäure, neigt besonders stark zur Polykondensation. Bereits Ansäuern einer alkalischen Silicat-Lösung führt zur Bildung von Polykieselsäuren, die in Abhängigkeit von den Reaktionsbedingungen als Sol (freibewegliche Partikel aus einigen hundert und mehr Atomen) oder als Gel (Netzwerk aus bindenden Wechselwirkungen zwischen den Partikeln) auftreten.
Versuch 24.2.9: Polykondensation von Kieselsäure, Tyndall-Effekt
Die Polykondensation führt dabei von linearen und cyclischen Polyanionen über bandförmige und flächige Kondensate zu dreidimensionalen Netzwerken. Ein Beispiel für ein Silicat, das sich von einer linear polykondensierten Kieselsäure ableitet, liegt im CaSiO3 vor.
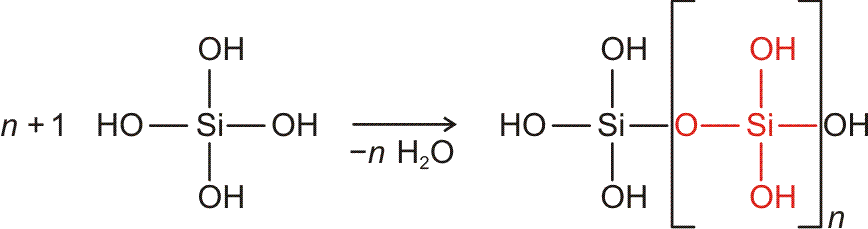
Die zugrunde liegende Säure, Metakieselsäure, hat die Summenformel H2SiO3, was der Zusammensetzung des Kettenbausteins in dem linearen Polymer entspricht. Die folgende Abbildung zeigt einen Ausschnitt aus einer Anionkette im CaSiO3. Dieses Calciumsilicat entsteht bei technischen Prozessen, bei denen eine Calciumkomponente des Rohstoffs mit Hilfe des Säureanhydrids SiO2 „verschlackt“ worden ist.
Ausschnitt aus der Metasilicat-Kette in CaSiO3.
Silicate sind in der Regel schwerlöslich in Wasser. Eine Ausnahme ist Natriummetasilicat, Na2SiO3, das wie CaSiO3 ein Salz der Metakieselsäure ist. Die wässrige Lösung („Wasserglas“) dient auch in der Technik als Ausgangsstoff für die Synthese von Silicaten wie den Zeolithen.
Obwohl die Silicate in einer Vielfalt auftreten, die von keinem anderen Element erreicht wird, haben die sich von den unterschiedlichsten Polykieselsäuren ableitenden Salze eine Gemeinsamkeit: das Grundmotiv der Kieselsäure, das SiO4-Tetraeder, bleibt in allen Polymeren erhalten. Darüberhinaus treten SiO4-Tetraeder stets eckenverknüpft auf, also weder kantenverknüpft noch flächenverknüpft. So liegen diese Merkmale auch bei einem weiteren Beispiel vor, das die Struktur eines Kupfersilicats mit einem flächigen Anion zeigt. Die Verbindung wurde bereits in der Antike als Blaupigment künstlich hergestellt: „Ägyptisch Blau“, CaCuSi4O10:
Ausschnitt aus der Kristallstruktur des Ägyptisch Blau, CaCuSi4O10. Farbcode: graugrün Si, blau Cu; Ca weggelassen.
Silicate, in denen ein Teil des Siliciums durch Aluminium ersetzt ist, heißen Alumosilicate. Durch die Substitution eines (vierbindigen) Si-Atoms durch eine (ebenfalls vierbindige) Al−-Gruppe erhalten die jeweiligen Gerüste eine höhere negative Ladung, das heißt, in den Verbindungen ist ein höherer Kationanteil enthalten. Beispiele sind Feldspäte wie NaAlSi3O8 und Zeolithe.
Zeolithe sind Alumosilicate, die sowohl in größerer Zahl in der Natur vorkommen, die aber auch in großem Umfang technisch hergestellt werden. Die allgemeine Formel ist
M+AlO2− · x SiO2 · y H2O
x kann in den Grenzen 1 (Zeolith A) bis unendlich variieren, M ist meist Natrium. Bei der Synthese der Zeolithe wird von Natriumaluminat- und Wasserglas-Lösungen ausgegangen.
Charakteristisches Strukturmerkmal der Zeolithe sind kanalartige Hohlräume in der Kristallstruktur, die die besonderen Eigenschaften der Zeolithe hervorrufen. Porendurchmesser und Porenvolumen sind so groß, dass für kleine Moleküle und Ionen die Möglichkeit gegeben ist, sich im kanalartigen Porensystem frei zu bewegen. Hieraus resultieren verschiedene Anwendungen von Zeolithen, vor allem als Katalysatoren und als „Ionenaustauscher“. So wird Zeolith A als kationreicher Zeolith (x = 1) in großem Umfang technisch hergestellt, um in Waschmitteln anstelle des Triphosphats zur Wasserenthärtung eingesetzt zu werden (siehe unten).
Während Triphosphat Störungen durch die Härtebildner des Wassers verhindert, indem es lösliche Verbindungen mit diesen eingeht, bindet Zeolith A diese Salze. Vor allem Ca2+-Ionen dringen in den Zeolith ein und werden dort gebunden, während eine äquivalente Menge Na+ in Lösung geht („Ionenaustausch“).
Die Abbildung zeigt schematisch den offenporigen Aufbau eines Zeoliths, dem Faujasit; dargestellt ist die Lage der Si-Zentren:
Um das Bauprinzip der sehr komplexen Struktur (hier ein bewegtes Bild der Elementarzelle) zu zeigen, sind nur die kürzesten Si···Si-Abstände als gelbe Linien dargestellt. Die kubische Struktur ist entlang der Raumdiagonale projiziert.
Die bisher angesprochenen Silicate sind kristalline Feststoffe. Aus Silicatschmelzen wie auch aus der Schmelze des reinen SiO2 können auch Gläser gewonnen werden.
Der Aufbau des Quarzglases ist typisch für den Glaszustand. Da beim Abkühlen der Schmelze kein Festpunkt überschritten wird, sondern die Flüssigkeit lediglich immer viskoser wird, entspricht auch der atomare Aufbau eines Glases dem einer Flüssigkeit und nicht dem eines kristallinen Feststoffes. So ergeben sich die typischen Merkmale eines Glases:
• Nahordnung um die Atome wie im Feststoff (bei Quarzglas: 4 O um Si, 2 Si um O), aber keine kristalline Fernordnung,
• kein Schmelzpunkt (Festpunkt), sondern allmähliches Zäherwerden der Schmelze; zwischen einer dünnflüssigen Schmelze und dem formstabilen Glaskörper liegt ein mehr oder weniger großer Temperaturbereich, in dem die Bearbeitung von Glas möglich ist.
Versuch 24.2.6: Temperaturwechselbeständigkeit von Quarzglas
Den im Versuch gezeigten nützlichen Eigenschaften des Quarzglases stehen für Anwendungen, bei denen die besonderen Eigenschaften dieses Werkstoffes keine Rolle spielen, Nachteile gegenüber: Hohe Temperaturbeständigkeit bedeutet auch, dass die Verarbeitung zu Geräten und Apparaturen nur bei hohen Temperaturen erfolgen kann (Knallgasbrenner).
„Glas“ im alltäglichen Sinne ist daher kein Quarzglas, sondern die Schmelze enthält neben Quarz verschiedene Metalloxide. Fensterglas enthält neben SiO2 noch Na2O (zugesetzt als Na2CO3, das gemäß Na2CO3 = „Na2O·CO2“ als Na2O-Quelle dient) und CaO (als CaCO3 zugesetzt, CaO („Branntkalk“, siehe unten) entsteht unter den Bedingungen der Glasschmelze). Der Zusatz von Metalloxiden zum Säureanhydrid SiO2 bewirkt Salzbildung:
≡Si–O–Si≡ + CaO → ≡Si–O− Ca2+ −O–Si≡
Durch die Salzbildung werden in das dreidimensionale SiO2-Netzwerk Trennstellen eingeführt. Die Ausdehnung vernetzter Bereiche nimmt hierdurch ab, der Schmelzbereich liegt bei tieferen Temperaturen.
Die Beschreibung dieser Salzbildung gelingt durch das Säure-Base-Konzept von Bjerrum, der eine Base als Spezies definiert, die ein Oxid-Ion, ein O2−-Ion auf eine Antibase überträgt. Bei der Reaktion von Calciumoxid und Siliciumdioxid ist dann CaO die Base und SiO2 die Antibase.
Das das Netzwerk aufbauende SiO2 wird als Netzwerkbildner bezeichnet, die die Trennstellen einfügenden Metalloxide als Netzwerkwandler. Netzwerkwandler werden gegenüber dem Netzbildner im Unterschuss eingesetzt, da bei äquimolaren Mengen Salze der Kieselsäure ohne Glaseigenschaften entstehen würden. Eine typische Glaszusammensetzung enthält daher weniger Baseanhydrid als zum Beispiel die erwähnten meta-Silicate: Na2O·CaO·6 SiO2 ist zum Beispiel angenähert die Zusammensetzung von „Natron-Kalk-Gläsern“ aus denen einfaches Flachglas und Behälterglas gefertigt wird.
Technische Gläser für spezielle Verwendungen enthalten wie einfaches Gebrauchsglas SiO2 als mengenmäßig wichtigsten Netzwerkbildner, ansonsten kann die Zusammensetzung jedoch stark variieren. So werden neben SiO2 als Netzwerkbildner eingesetzt: B2O3, P2O5, As2O5, auch Al2O3; Netzwerkwandler sind: Li2O, Na2O, K2O, MgO, CaO, BaO, ZrO2, PbO2, auch Al2O3.
Die Verarbeitungstemperatur liegt je nach der Glaszusammensetzung bei ca. 800 bis 1400 °C, also deutlich unter der für die Bearbeitung von Quarzglas notwendigen Temperatur (ca. 2000 °C). Die so erhaltenen Gläser weisen eine hohe Druckfestigkeit auf. Kritischere Größen sind Biege- und Zugfestigkeit, die von der Güte der Glasoberfläche abhängen. Ein wirksames Verfahren zur Erhöhung der Biege- und Zugfestigkeit von Gläsern ist die Erzeugung von Druckspannung an der Glasoberfläche. Hierzu werden zwei Verfahren eingesetzt: (a) Die thermische Härtung: Der Glasgegenstand wird bis nahe an die Erweichungstemperatur erhitzt, dann werden die oberflächennahen Bereiche abgeschreckt, indem Luft angeblasen wird oder in eine Flüssigkeit eingetaucht wird (Beispiel: Sekurit-Glas für Autoscheiben); (b) die chemische Härtung: Beim Eintauchen natriumhaltiger Gläser in eine Kaliumsalzschmelze (KNO3) werden an der Oberfläche des Glases Natrium-Ionen durch die größeren Kalium-Ionen ersetzt; es bildet sich eine Druckspannung aus (Anwendung: Brillengläser).
Versuch 24.2.13: Gläser zeigen
Die Regel, dass Silicate stets aus eckenverknüpften SiO4-Tetraedern aufgebaut sind, gilt für die Entstehung unter Normaldruck. Sowohl von Silicaten als auch von SiO2 selbst sind Hochdruckmodifikationen bekannt, denen bei geologischen Prozessen im Erdinnern eine wichtige Rolle zugeschrieben wird. Die Strukturen enthalten dann fünf- oder sechsfach koordiniertes Silicium. Ein Beispiel ist die Struktur der Hochdruckform des CaSiO3, die im Perowskit-Typ kristallisiert (der Strukturtyp hat seinen Namen vom Mineral Perowskit, CaTiO3):
Eine natürlich vorkommende Hochdruckform des SiO2 ist der Stishovit, der in der Rutilstruktur kristallisiert und der erstmals im Nördlinger Ries gefunden wurde (?).
Im Labor bildet sich oktaedrisch koordinertes Silicium beim analytischen Silicat-Nachweis bei der „Wassertropfenprobe“. Hierbei wird HF und SiO2 zu SiF4 umgesetzt und dieses anschließend wieder hydrolysiert. Unter besonderen Bedingungen kann dabei auch [SiF6]2− gebildet werden, das Hexafluoridosilicat. Ein schwerlösliches Salz mit diesem Anion ist Ba[SiF6]. Beim Erhitzen zerfällt dieses zu BaF2 und SiF4 – ein Weg, um reines SiF4 herzustellen. Der Zerfall illustriert eine in Teil A beschriebene Regel: es werden so viele dative Si-F-Bindungen heterolytisch gespalten, bis das ungeladene Molekül SiF4 entstanden ist; Zufuhr der Bindungsdissoziationsenergie würde dann erst zum homolytischen Bruch normaler kovalenter Si-F-Bindungen führen.
Die Chemie des Germaniums ist derjenigen des Siliciums sehr ähnlich, außer dass es aufgrund seiner etwas größeren Atom- und Ionenradien häufiger in einer höheren Koordinationszahl als vier auftritt.
Zinn kristallisiert in zwei polymorphen Formen. Das bei Raumtemperatur beständige „weiße“ oder β-Zinn wandelt sich unterhalb von ca. 13 °C sehr langsam in feinpulvriges „graues“ oder α-Zinn um („Zinnpest“). Die Dichte der stabilen α-Modifikation ist erheblich geringer als diejenige des β-Allotrops (5,77 gegenüber 7,29 g cm−3). Damit übereinstimmend ist die Koordinationszahl im weißen Zinn höher als im grauen. Das folgende Bild zeigt den strukturellen Zusammenhang zwischen den beiden Modikationen. Graues Zinn kristallisiert in der Diamantstruktur. Die β-Zinn-Struktur entsteht nun durch eine ungewöhnlich drastische Kompression entlang einer der Basisvektoren der kubischen Elementarzelle.
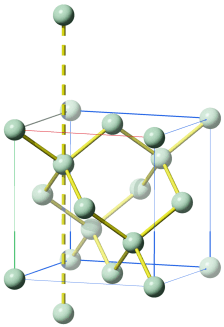
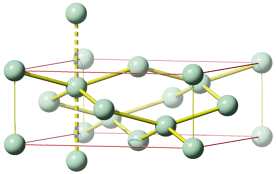
Nach der Kompression sind zu den vier Kontakten zu den nächsten Nachbarn der Diamantstruktur zwei neue Kontakte hinzugekommen und zwar zu den beiden Atomen, die dem betrachteten in den beiden angrenzenden Elementarzellen in Kompressionsrichtung translationsäquivalent sind (gestrichelte Linien in den Abbildungen). Man beachte, dass jedes Zinnatom im unteren Bild zu zwei zusätzlichen Nachbarn bindet, was aber der Übersichtlichkeit halber nur für ein Sn-Atom gezeigt ist.
Zinn neigt beim Abkühlen aus der Schmelze ungewöhnlich stark dazu, große einkristalline Bereiche im erstarrten Zustand auszubilden. Beim Verbiegen von Zinnstangen, die durch Ausgießen der Schmelze gewonnen wurden, ist daher deutlich das Aneinanderreiben der großen kristallinen Bereiche zu hören – das „Zinngeschrei“.
Versuch 24.3.?: Zinngeschrei
Bei stabilen Kohlenstoff-, Silicium- und Germanium-Verbindungen wird von einer Beteiligung von s- und p-Orbitalen an den Bindungen ausgegangen. Halogenide dieser Elemente in der Oxidationsstufe +II sind daher reaktiv, zum Beispiel das bei organischen Synthesen genutzte CCl2 („Dichlorcarben“), das durch HCl-Abspaltung aus CHCl3 durch starke Base erzeugt werden kann. Zinn dagegen zeigt eine ausgeprägte Chemie in den beiden Oxidationsstufen +II und +IV. Es deutet sich der bei Blei stark ausgeprägte inert-pair-Effekt an. Hierunter wird verstanden, dass sich bei schweren Elementen das s-Elektronenpaar der Valenzschale nur eingeschränkt an Bindungen beteiligt. Dementsprechend wird Zinn durch das schwache Oxidationsmittel H3O+ nur zur zweiwertigen Form oxidiert, stärkere Oxidationsmittel wie Iod oder Sauerstoff führen jedoch zu Zinn(IV).
Versuch 24.3.?: Auflösen von Zinn in konz. Salzsäure
• Sn + 2 HCl → SnCl2 + H2
Versuch 24.3.?: Zinn(II)-chlorid-Lösung entfärbt Iod
• SnCl2 + I2 + 4 Cl− → [SnIVCl6]2− + 2 I−
Versuch 24.3.?: Zinn verbrennt an Luft
• Sn + O2 → SnO2
Während Zinn(II)-chlorid (Zinndichlorid) sich in seinen Eigenschaften einem typischen Metallchlorid annähert, zum Beispiel dissoziiert es in Wasser unter Bildung hydratatisierter Kationen – die aber doch recht stark acide sind (pKS = 1,9), ist Zinn(IV)-chlorid (Zinntetrachlorid) ein typisches Säurechlorid, das in Wasser hydrolysiert.
Versuch 24.3.?: Hydrolyse von Zinn(IV)-chlorid
• SnCl4 + 4 H2O → Sn(OH)4 + 4 HCl
Das Verhalten von Zinn folgt einer allgemeinen Regel: je höher die Oxidationszahl eines Elements, umso saurer ist dessen Hydroxid oder dessen Aquakomplex im Sinne Brønsteds, umso saurer ist dessen Oxids im Sinne Bjerrums. Man vergleiche hierzu auch die Abstufung der Säurestärken in Reihen wie HClO, HClO2, HClO3, HClO4; H2SO3, H2SO4; HNO2, HNO3, bei denen die höhere Oxidationsstufe des Zentralelements stets die höhere Acidität aufweist.
Blei ist in jeder Hinsicht ein typisches Metall. Es kristallisiert in der kubisch-flächenzentrierten Struktur mit der hohen Koordinationszahl 12, es leitet den elektrischen Strom, es ist duktil und zeigt den charakteristischen Metallglanz. Das Zusammentreffen dieser Eigenschaften geht mit dem metalltypischen Mangel an Elektronen einher – ein Mangel, der erstaunlich ist, Blei hat schließlich dieselbe Valenzelektronenzahl wie Kohlenstoff. Es zeigt sich hier deutlich, dass für die Bindung im elementaren Blei eben nicht vier Elektronen zur Verfügung stehen, sondern wegen des inert-pair-Effekts nur zwei. Die Chemie des Bleis stimmt mit diesem Befund überein. Blei(II)-Verbindungen sind stabil, Blei(IV)-Verbindungen sind – falls sie überhaupt existieren – meist starke Oxidationsmittel.
Auch die Herstellung von Bleimetall aus seinen Erzen folgt dem Standardverfahren für zahlreiche Gebrauchsmetalle, nämlich der Reduktion des Oxids durch Kohle:
Versuch 24.3.?: Bleiherstellung aus Blei(II)-oxid und Kohle
• PbO + C → Pb + CO
Von rechts nach links durch das Periodensystem zu gehen heißt, zunehmend Elektronenmangel kennen zu lernen. In der 13. Gruppe kann zum ersten Mal nicht ohne weiteres die Oktettregel erfüllt werden. Aluminium und seine schweren Homologen reagieren darauf normal: die Elemente sind Metalle, die Verbindungen werden ionischer. Bor aber ist einzigartig: Elektronenmangel + Kovalenz führen zu einer Chemie, die kaum eine Parallele bei anderen Elementen findet.
Versuch 25.1.1: Ikoseader aufstellen
Die Modifikationen von reinem und etwas verunreinigtem (wenige % C oder N) Bor zeigen einen gemeinsamen und für Bor charakteristischen Baustein und zwar B12-Ikosaeder (Ikosaeder = Zwanzigflächner).
(Noch ein Test mit einem bewegten Bild .)
Benachbarte Ikosaeder sind durch B-B-Bindungen miteinander verbunden, was zu dreidimensional kovalent vernetzten Strukturen führt. Ähnlich wie der dreidimensional kovalent vernetzte Diamant ist elementares Bor sehr hart. Die ikosaedrischen Bausteine sind Ausdruck einer Sonderstellung des Bors im Vergleich zu allen anderen Elementen: die Regel „Elektronenmangel = metallische Eigenschaften“ gilt bei Bor nicht. Stattdessen kommt es zu einer „kovalenten Alternative“ zur Metallbindung, nämlich zu 2-Elektronen-Mehrzentren-Bindungen. Die kompliziert aufgebauten Strukturen von elementarem Bor und borreichen Verbindungen sind zur Erklärung solcher Bindungen weniger gut geeignet als kleine Moleküle, die nach demselben Prinzip aufgebaut sind: die Borane.
BH3, die auf dem Papier einfachste Verbindung zwischen Bor und Wasserstoff, ist nur wenig stabil. In Abwesenheit von Reaktionspartnern bildet sie ein Dimer, das Diboran:
Bei Versuchen, eine Lewis-Formel des Diborans unter Verwendung von 2e-2z-Bindungen zu formulieren, werden keine Elektronenoktetts für die Boratome erhalten. Auch die Valenzbindungsmethode lässt daher für Borane und verwandte Verbindungen 2-Elektronen-Mehrzentrenbindungen zu, und zwar 2e-3z-Bindungen, bei denen jeweils zwei Elektronen jedem B-H-B-Fragment zugeordnet werden.
Da die MO-Methode von der Grundidee her eine Delokalisation von Elektronenpaaren über das gesamte Molekül vorsieht, stellt sich Diboran bei dieser Beschreibung als weniger aus der Reihe fallend dar. Die Orbitale des folgenden Schemas werden erhalten, indem bei jedem Boratom die vier Orbitale 2s, 2px, 2py und 2pz zu MOs kombiniert werden, und anschließend die vier 1s-Orbitale der Wasserstoffatome bindend an die MOs angefügt werden. Dabei ist die Abfolge der aus den p-Orbitalen gebildeten MOs auf der Energieskala ein Rechenergebnis, das nicht durch naheliegende Betrachtungen abzuleiten ist. Die Abbildung zeigt nur die besetzten sechs Valenzorbitale (6 e− von 2 B + 4 e− von 4 H = 6 Elektronenpaare). Das höchste besetzte Orbital (HOMO) leitet sich von einer antibindenden π-Wechselwirkung zwischen den Boratomen ab.
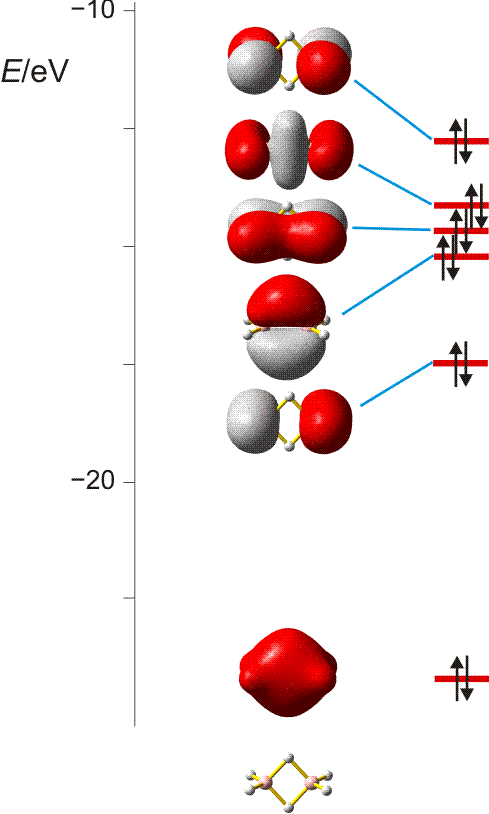
Die Beiträge der Bor-Atomorbitale in derselben energetischen Reihenfolge:
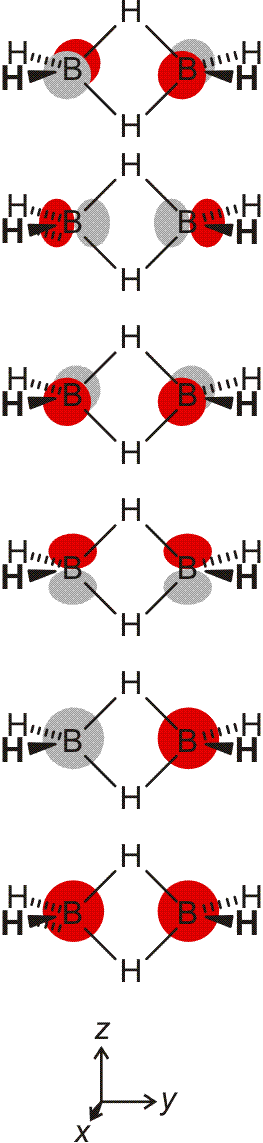
Borane sind Lewis-Säuren, die ihre 2e-3z-Bindungen aufgeben, wenn Elektronenpaardonoren die Bildung von 2e-2z-Bindungen erlauben. So reagieren Alkalimetallhydride mit Boranen zu Boranaten (Hydridoboraten). Das einfachste Boranat ist das [BH4]−-Anion, das große Bedeutung als Reduktionsmittel, vor allem in der organischen Synthese, besitzt.
Versuch 25.1.2: Natriumboranat als Reduktionsmittel
Auch die trigonal-planar aufgebauten Borhalogenide sind starke Lewis-Säuren. Erstaunlicherweise ist BF3 nicht – wie erwartet – die stärkste Lewissäure in der Reihe der Borhalogenide. Die Ursache hierfür ist, dass die Elemente der zweiten Periode, Bor und Fluor, den Elektronenmangel am Boratom durch pπ-pπ-Bindungen abschwächen können.
Die Atomradien der Elemente nehmen in einer Periode mit steigender Ordnungszahl ab, in einer Gruppe nehmen sie mit der Hauptquantenzahl zu. Ähnliche Radien ergeben sich wegen dieser beiden Trends für ein Element und dessen Nachbarn schräg rechts darunter. Zwei Elemente in Paaren wie zum Beispiel Li/Mg, Be/ Al, B/Si oder C/P weisen daher oft gewisse Gemeinsamkeiten auf. Dieser Umstand wird Schrägbeziehung genannt.
Das Paar B/Si zeigt eine Reihe solcher Gemeinsamkeiten, so ist die Struktur von B2O3 wie die Tridymitstruktur chiral, da Boroxid-Schrauben vorliegen. Beim Auflösen in Wasser reagiert Boroxid als Säureanhydrid, es entsteht Borsäure, H3BO3. Diese zeigt nun sowohl Gemeinsamkeiten mit Kohlensäure als auch mit Kieselsäure. In den farblosen Kristallen von Borsäure liegen planare H3BO3-Moleküle vor, die eine dreizählige Symmetrieachse aufweisen:
Der Aufbau der Borsäure entspricht damit demjenigen der Kohlensäure – mit der Ausnahme, dass nicht die Oktettregel gilt, wenn nicht analog zu BF3 eine B-O-π-Bindung formuliert wird.
Borsäure ist eine sehr schwache Säure. Durch Zusatz starker Base (NaOH, etc.) gibt sie formal ein Proton ab. Die konjugierte Säure, ein Hydrogenborat der Formel H2BO3−, das analog zu Kohlensäure mit einer B=O-Doppelbindung zu formulieren wäre, ist jedoch nicht bekannt. Stattdessen liegt in Kristallen des Mononatriumsalzes der Borsäure ein Tetra(hydroxido)borat-Ion, [B(OH)4]−, vor, das wie die Orthokieselsäure aufgebaut ist:
Weiterhin im Gegensatz zu Kohlensäure und analog zur Kieselsäure neigt Borsäure zur Polykondensation und bildet dabei kristalline und glasartige Polyborate. Eines der wichtigsten Borminerale ist Borax, Na2B4O5(OH)4·8 H2O (man findet auch die weniger aussagekräftige Formel Na2B4O7·10 H2O). Im Tetraborat-Ion des Borax treffen die verschiedenen Bauelemente zusammen: siliciumtypische BO4-Tetraeder, aber auch analog zur Kohlensäure planare, dreifach koordinierte Boratome:
Versuch 25.1.5: Boraxperlen
Eine besonders charakteristische Reaktion, mit der Borsäure zuverlässig identifiziert werden kann, ist die Verbrennung des Trimethylesters, der aus Borsäure und Methanol säurekatalysiert gebildet wird.
Versuch 25.1.3: Borsäuretrimethylester
• B(OH)3 + 3 MeOH → B(OMe)3 + 3 H2O (Me = CH3)
Die Esterbildung gelingt auch mit mehrwertigen Alkoholen („Polyolen“). Da die Bildungstendenz der tetraedrischen Anionen größer als die von Tetra(hydroxido)borat ist („Chelateffekt“), erscheint die Borsäure saurer.
Versuch 25.1.4: Titration von Borsäure
Bei der Umsetzung Lewis-acider Borverbindungen mit Ammoniakderivaten entstehen Lewis-Säure-Base-Addukte, von denen das Amminboran in Teil A erwähnt wurde. Die Amminboran-Struktur erschließt sich vom Ethan ausgehend, indem ein C2-Fragment durch ein isoelektronisches BN-Paar ersetzt ist. Dieses Prinzip ist nicht auf Amminboran beschränkt, vielmehr gibt es weitere Analoge organischer Verbindungen wie zum Beispiel das Borazin B3N3H6, das in enger Beziehung zum Benzol steht.
Besonders interessant ist in diesem Zusammenhang das Bornitrid, BN, das isoelektronisch mit dem Kohlenstoff ist. Tatsächlich sind zwei Modifikationen bekannt, die dem Graphit und dem Diamant entsprechen und wichtige Werkstoffe sind; ein B30N30 fehlt allerdings bislang.
Oxidierbar wie Magnesium, das Hydroxid amphoter wie Sb(OH)3 – die Chemie des Aluminiums scheint sich aus den thermodynamischen Basisgrößen zu ergeben: dem niedrigen Standardpotential und der recht hohen Acidität des Hexaaquaaluminium-Ions. Dass Aluminium nach Eisen das bedeutendste Gebrauchsmetall ist, kann bei diesen Randbedingungen kaum vermutet werden. Die besonderen Eigenschaften des Aluminiumoxids aber ändern alles.
Zuerst die Amphoterie des Hydroxids, die mit der Acidität von Aluminiumsalzen einhergeht. Der amphotere Charakter von Al(OH)3 ermöglicht die Trennung von Aluminiumerzen von Verunreinigungen durch Eisen(III)-(hydr)oxid.
Versuch 25.2.9: Acidität von Aluminiumsalzen
Versuch 25.2.1: Trennung von Aluminium und Eisen
• Al(OH)3 + OH− ⇌ [Al(OH)4]−
Die elektrolytische Herstellung von Aluminium wird im Kapitel „Gewinnung von Metallen“ beschrieben. Das so gewonnene unedle Metall sollte bei jedem pH-Wert von H+I oxidiert werden.
Versuch 25.2.5: Aluminium löst sich in Säure und Lauge, nicht aber in Wasser
• 2 Al + 6 H3O+ → 2 [Al(H2O)6]3+ + 3 H2 + 6 H2O
• 2 Al + 6 H2O + 2 OH− → 2 [Al(OH4)]− + 3 H2
Versuch 25.2.3: Aufhebung der Passivierung von Aluminium durch Quecksilber oder Gallium
Korund, Al2O3, bildet eine Struktur, die als Ionenkristall mit ungewöhnlich hoher Gitterenergie gesehen werden kann. Die hohe Gitterenergie hat zwei Ursachen: die Struktur hat eine hohe Raumerfüllung (eine große Madelung-Konstante) und die Ladung der Ionen ist hoch. Die Korundstruktur beruht auf einer hexagonal dichtesten Packung von Oxid-Ionen. Die Al3+-Kationen besetzten Oktaederlücken und zwar sind die Lücken aller Oktaederlückenschichten zu 2/3 belegt. Aus einer dichtest gepackten Schicht lückenfüllender Atome wird so ein Wabenmuster. Die folgende Abbildung zeigt drei Lagen von Oxid-Ionen (große rote Kugeln) mit Aluminium-Kationen in Oktaederlücken (kleine graue Kugeln)
Ein Schwerpunktmodell zeigt die Verteilung der Aluminium-Ionen deutlicher; man beachte die für viele Metall(III)-oxide und -hydroxide typische Kantenverknüpfung von MIIIO6-Oktaedern.
Die hohe Stabilität der Korundstruktur zeigt sich unmittelbar in der hohen Standardbildungsenthalpie des Al2O3 von −1670 kJ mol−1:
Versuch 25.2.7: Exotherme Bildung von Aluminiumoxid
Die hohe Exothermie der Korundbildung ist Grundlage des Thermitverfahrens, mit dessen Hilfe kleine Mengen beliebig zusammengesetzter Stahlschmelzen aluminothermisch hergestellt werden können, zum Beispiel für das stromlose Verschweißen von Schienen oder im Brückenbau. Man beachte die spezielle Reaktionsführung beim Thermitversuch: eine Aktivierung der stark exothermen Reaktion durch Aufheizen des gesamten Gemisches würde zu dessen Explosion führen!
Versuch 25.2.8: Thermit
Neben dieser Spezialanwendung werden einige Metalle aluminothermisch hergestellt (siehe Kapitel „Gewinnung von Metallen“).
Aluminiumhydroxid ist amphoter. Damit übereinstimmend kann auch Aluminiumoxid als Säureanhydrid oder als Baseanhydrid reagieren, es kann in Schmelzen also im Sinne Bjerrums als Base oder als Antibase reagieren. Al2O3 wirkt als Base, wenn es in Säuren oder in Schmelzen von Säureanhydriden unter Bildung von Aluminiumsalzen aufgelöst wird. Die Koordination der Aluminiumatome ist in Salzen wie dem Sulfat derjenigen in Korund ähnlich: die Al-Atome sind oktaedrisch sechsfach von O-Atomen koordiniert.
Wird Korund dagegen mit einem stärkeren Baseanhydrid umgesetzt, so entstehen Aluminate, deren Aufbau große Ähnlichkeit mit Silicaten zeigt. Das folgende Bild zeigt die Struktur eines CaAl2O4, das formal als CaO·Al2O3 abgeleitet werden kann. Die Strukturanalyse zeigt tetraedrische Umgebung de Al-Atome sowie Eckenverknüpfung der einzelnen AlO4-Tetraeder. Da jedes O-Atom auf diese Weise zwei Al-Atome verknüpft, ist die Formel des gebildeten anionischen Netzwerks (AlO4/2)− = (AlO2)−. Es stimmen also Formeltyp, die Umgebung der einzelnen Atome und die Art der Verknüpfung mit SiO2 überein. Die Ladung des Aluminat-Netzwerks wird von Calcium-Ionen kompensiert: Ca½(AlO2) = CaAl2O4.
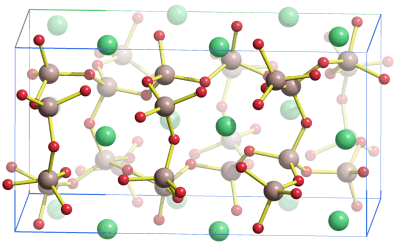
Die Kristallstruktur des Calciumaluminats CaAl2O4. Farbcode: beige-braun Al, grün Ca.
Bei den aluminiumhalten Silicaten ist der besonders interessante Fall bekannt, dass Aluminiumoxid gegenüber ein und demselben Säureanhydrid, nämlich SiO2, sowohl als Säure- wie auch als Baseanhydrid auftreten kann. So sind Tone Aluminiumsilicate, in deren Struktur ein anionischer Silicatteil identifizierbar ist, dessen Ladung von Al3+-Ionen ausgeglichen wird. Dieses Prinzip findet sich auch im Mineral Andalusit:
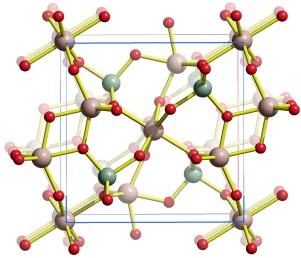
Die Kristallstruktur des Andalusits. Farbcode: beige-braun Al, graugrün Si.
Die Summenformel des Andalusits ist Al2SiO5, formal in Säure- und Baseanhydrid aufgeteilt also Al2O3·SiO2. In der Struktur ist jedes Siliciumatom von vier Sauerstoffatomen umgeben, wobei keine Si-O-Si-Verbrückungen auftreten. Ferner verbrückt eines der fünf O-Atome der Summenformel drei Aluminiumatome, hat jedoch keinen Kontakt zu Silicium. Man könnte dies in der Formel Al2O(SiO4) ausdrücken, einem Aluminiumoxid-orthosilicat.
Ist Aluminiumoxid nicht die stärkste Bjerrum-Base in der Schmelze der Komponenten, so kommt es zu der oben gezeigten Aluminat-Variante. So enthalten Feldspäte die zu Aluminium äquivalente Menge an Alkali-Kationen. Aluminium kann damit als (AlO2)− nicht nur formal als Baustein eines SiO2-analogen Gerüst angesehen werden, sondern es bilden sich in der Realität gemischte anionische Al/Si-Netzwerke aus. Als Beispiel ist eine Form des Kalifeldspats abgebildet und zwar das Mineral Mikroklin:
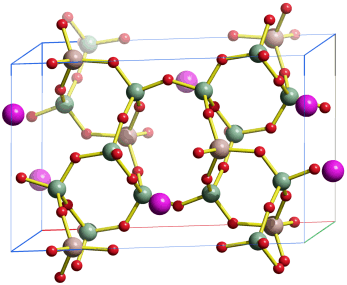
Die Kristallstruktur des Feldspats Mikroklin, KAlSi3O8. Farbcode: beige-braun Al, graugrün Si, violett K.
Feldspäte, bei denen Al-Atome einzelne Si-Atome in einem Silicat-Netzwerk ersetzt haben, gehören zu den Alumosilicaten. Die Unterscheidung zwischen Alumosilicaten (Al im anionischen Netzwerk) und Aluminiumsilicaten (Al als Gegenion eines Silicatnetzwerks) führt natürlich zu der Frage, wie beim Betrachten einer Struktur mit Al- und Si-Atomen entschieden werden kann welcher Typ nun vorliegt. Beachten Sie folgende Regel: Alumosilicate enthalten tetraedrisch koordinierte Al-Atome, bei Aluminiumsilicaten ist die Koordinationszahl der Al-Atome größer (bei Andalusit fünf und sechs).
Auch in der 13. Gruppe ist der inert-pair-Effekt wirksam. Während die in den letzten Jahren intensiv untersuchten Aluminium(I)-, Gallium(I)- und Indium(I)-Verbindungen zwar strukturell faszinierende Metallcluster aufbauen, verlangt deren hohe Neigung zur Oxidation und zur Disproportionierung aufwendige synthetische Methoden. Bei Thallium hingegen ist wie bei Blei(II) und Bismut(III) die Oxidationsstufe die stabilste, die um zwei Einheiten unter der Valenzelektronenzahl liegt. Thallium(I)-Verbindungen weisen dabei Ähnlicheit sowohl zu den Alkalielementen (TlOH ist ein stark basisches Hydroxid) als auch zu Silber(I) auf (TlCl ist schwerlöslich). Beim Umgang mit Thalliumsalzen ist deren hohe Giftigkeit zu beachten: Thallium(I)-sulfat ist das klassische, heute aber ungebräuchliche Rattengift.
Während Berylliumhydroxid noch amphoter ist, leiten sich von den schwereren Erdalkalielementen stark basische Verbindungen ab. Calciumoxid ist das Anhydrid der starken Base Calciumhydroxid. In zahlreichen technischen Prozessen dient CaO als basische Komponente bei der Reaktion mit Säureanhydriden wie SiO2 und auch Al2O3.
Versuch 26.1: Mineralien aufstellen
Das mengenmäßig wichtigste Erdalkalielement ist Calcium. Calcium ist deutlich unedler als Aluminium. Da Calcium(hydr)oxid keine wirksam passivierenden Schichten aufbaut, reagiert Calcium sogar mit Wasser:
Versuch 26.3: Reaktion von Calcium mit Wasser
• Ca + 2 H2O → Ca(OH)2 + H2

Calciumcarbonat (Kalkstein, Kreide, Marmor) und auch das Doppelsalz Dolomit, CaMg(CO3)2, bilden ganze Gebirge. CaCO3 kommt in mehreren Modifikationen vor; die bei Raumtemperatur stabile Form ist Calcit, nur wenig unstabiler ist Aragonit, deutlich unstabiler ist Vaterit. Alle drei Formen werden von Organismen zum Aufbau von Schalen verwendet, Muscheln bauen zum Beipiel sehr beständige Schalen auf, in denen Calcit und Aragonit miteinander abwechseln. Beim Menschen machen kleine Calcitkristalle unser Gleichgewichtsorgan im Innenohr möglich.
Calciumcarbonat, CaCO3, ist in Wasser schwerlöslich (L = 2,25 · 10−8). Trotzdem löst sich Kalkstein in der Natur langsam in Wasser auf; der Grund hierfür liegt im Kohlensäuregehalt des Wassers, der durch die Aufnahme von CO2 aus der Luft resultiert. CO2-haltiges Wasser löst Calciumcarbonat nämlich unter Bildung von Calciumhydrogencarbonat.
Versuch 24.1.11/26.6: Fällen und Wiederauflösen von Kalk
• Ca(OH)2 + CO2 → CaCO3 + H2O
• CaCO3 + H2O + CO2 → Ca(HCO3)2
Es handelt sich um eine Gleichgewichtsreaktion, die dann von rechts nach links abläuft, wenn dem Gleichgewicht eine Komponente entzogen wird. So führt das Entweichen von Kohlendioxid zum Ausfallen von CaCO3: Beim Eindunsten von Calciumhydrogencarbonat-Lösungen bildet sich CaCO3 (Bildung von Stalagmiten und Stalagtiten in Tropfsteinhöhlen), und beim Erhitzen von Calciumhydrogencarbonat-Lösungen fällt ebenfalls CaCO3 aus (Kesselstein).
Calciumsalze zählen gemeinsam mit Magnesiumsalzen zu den Härtebildnern des Wassers. Die Härte des Münchner Trinkwassers entfällt zu mehr als 3/4 auf dessen Calciumgehalt, der Rest auf Magnesiumsalze. Die für Münchner Trinkwasser angegebenen Härtewerte von 15,2 °dH Gesamthärte und 13,8 °dH Carbonathärte sind typisch für ein Wasser, das Kontakt mit einem Kalkgebirge hatte (1 °dH entspricht 1 mg CaO pro 100 mL; hierzu wird der Calcium- und Magnesiumgehalt auf CaO umgerechnet; werden zum Beispiel 20 mg L−1 Magnesium ermittelt, so tragen diese 2 mg pro 100 mL = 2/24,3 mmol pro 100 mL = 0,0823 mmol pro 100 mL = 0,0823 · 56,1 mg CaO pro 100 mL = 4,6 mg CaO pro 100 mL mit 4,6 °dH zur Gesamthärte bei). Die Anionen der härtebildenden Salze sind vor allem Hydrogencarbonat und Sulfat. Da gemäß der Reaktion bei der Kesselsteinbildung die durch den Ca(HCO3)2-Anteil hervorgerufene Härte durch Erhitzen beseitigt werden kann, heißt diese temporäre oder vorübergehende Härte oder Carbonathärte, während der als Sulfat vorliegende Anteil (= Gesamthärte − Carbonathärte) als bleibende oder permanente Härte bezeichnet wird.
Der Calcium- und Magnesiumgehalt von Wasser ist für die Ernährung wichtig, führt aber zu zahlreichen technischen Problemen. Für viele Zwecke wird daher Wasser benötigt, das frei von Härtebildnern ist. Das heute übliche Verfahrung zur Wasserenthärtung ist der Ionenaustausch.
Versuch 26.7: Calcium als Härtebildner
Versuch 26.8: Ionenaustauscher
Calciumoxid ist die billigste Base der Technik. Es wird neben Kohlendioxid durch das „Brennen“ (Erhitzen auf 900–1100 °C) von Kalkstein gewonnen. Das gewonnene CaO reagiert stark exotherm mit Wasser zu Calciumhydroxid („Löschkalk“); festes Ca(OH)2 wird gewonnen, indem noch im Brennofen zum Branntkalk die äquimolare Menge Wasser zugegeben wird.
Versuch 26.4: Kalkbrennen
• CaCO3 → CaO + CO2
Versuch 26.5: Kalklöschen
• CaO + H2O → Ca(OH)2
Kalkmörtel ist ein Gemisch aus Kalkbrei und Sand. Kurze Zeit nach der Verarbeitung bindet Mörtel ab, das heißt überschüssiges Wasser tritt aus, es entsteht ein trockenes Gemisch aus Ca(OH)2 und Sand. Beim anschließenden Erhärten, das sich bei dickem Mauerwerk über Jahrzehnte oder sogar Jahrhunderte erstrecken kann, reagiert das Calciumhydroxid mit dem Kohlendioxid der Luft zu Calciumcarbonat.
Kalkmörtel ist ein Luftmörtel, das heißt, es findet weder ein Festwerden unter Wasser statt noch ist der abgebundene Mörtel wasserfest. Ein auch unter Wasser abbindender und wasserfester Mörtel, hydraulischer Mörtel, lässt sich unter Verwendung von Portland-Zement herstellen. Der Ausgangsstoff, Portland-Zement-Klinker, entsteht, wenn nicht Kalkstein allein, sondern Kalkstein im Gemisch mit Ton (Aluminiumsilicat) – oft als natürlich vorkommendes Gemisch („Mergel“) – auf ca. 1450 °C erhitzt wird. Es entstehen Calciumalumosilicate, deren Zusammensetzung meist im Sinne formaler Komponenten angegeben wird. Diese Komponenten sind vor allem Tricalciumsilicat, „3 CaO·SiO2“, und Calciumaluminate, zum Beispiel „3 CaO·Al2O3“. Ein typischer Zementklinker enthält ca. drei Teile Silicat- und ein Teil Aluminatkomponente. Zur Herstellung von Portland-Zement wird dem Klinker Gips (Calciumsulfat) als Erstarrungsverzögerer zugefügt. Das Erstarren von Zement ist eine Hydratation. Dabei entstehen wasser- und sulfathaltige Calciumalumosilicate, deren Kristalle miteinander verfilzt sind, woraus sich die große Festigkeit von erhärteten Zement-Produkten ergibt (Beton).
Zementklinker anstelle wertloser Abfallstoffe herzustellen ist das Ziel zahlreicher technischer Verfahren. Zu den sich besonders schnell anhäufenden nutzlosen Abfallprodukten zählt „Chemiegips“, das ist Calciumsulfat, das bei der Phosphatherstellung, aber auch bei der Rauchgasentschwefelung anfällt (2 SO2 + 2 Ca(OH)2 + O2 + 2 H2O → 2 CaSO4·2 H2O). In der Natur kommt Calciumsulfat ebenfalls als Dihydrat, Gipsstein, CaSO4·2 H2O, vor. Wasserfreies Calciumsulfat kommt als Anhydrit, CaSO4, als bei Wasserkontakt quellbare Gesteinsschicht vor, deren Volumenvergrößerung bei der Hydratation zu Gipsstein fatale geologische Folgen beim Tunnelbau und anderen Bohrungen nach sich zieht. Beim Erhitzen von Gipsstein auf 120–180 °C wird das Halbhydrat, CaSO4·½ H2O erhalten, das als Baustoff verwendet wird („Stuckgips“ und „Estrichgips“). Chemiegips ist für diesen Zweck aufgrund seiner mangelhaften Abbindeeigenschaften ungeeignet. Ein Verfahren zur Verwertung von Chemiegips benutzt gleich eine ganze Reihe von Grundregeln chemischer Reaktionsführung, die in dieser Vorlesung behandelt wurden. Umgesetzt werden: Chemiegips + Koks + Ton (Aluminiumsilicat), erhalten wird SO2 für die Schwefelsäureproduktion und Zementklinker als Baustoff. Erkennen Sie die angewandten Gesetzmäßigkeiten?
Calciumoxid enthält die sehr starke Base O2−. Die Fähigkeit des Calciums, stabile Ionenstrukturen zu bilden, wird genutzt, um weitere sehr reaktive Anionen handhabbar zu machen. So enthält das als Düngemittel hergestellte Calciumcarbodiimid CN22−-Ionen, die beim Kontakt mit Wasser doppelt protoniert werden und dabei Cyanamid, NC-NH2 bilden, welches wiederum zu Harnstoff und dessen Hydrolyseprodukten weiterreagiert.
Versuch 26.10: Hydrolyse von Calciumcarbodiimid (Kalkstickstoff)
• CaCN2 + 2 H2O → Ca(OH)2 + H2N-CN
Calciumhydrid enthält die ebenfalls sehr starke Base Hydrid, die im Na[BH4] durch polare Atombindungen an Bor gebunden und damit in ihrer Reaktivität gedämpft vorlag. In der ionischen CaH2-Struktur ist die Reaktivität erheblich höher.
Versuch 26.13: Hydrolyse von Calciumhydrid
• CaH2 + 2 H2O → Ca(OH)2 + 2 H2
Die Elemente am linken Rand des Periodensystems haben nur noch eins im Sinn – ihr Valenzelektron abzugeben und als einwertige Kationen Bindungen mit starkem ionischen Charakter aufzubauen. „Schwerlöslichkeit“ wird in der 1. Gruppe zum Fremdwort. Viele Eigenschaften von Anionen in Lösung – Hydroxid, Hydrogencarbonat, Carbonat, Phosphat – lassen sich erst mit den Alkali-Ionen als Gegenionen studieren, da viele andere Kationen schwerlösliche Salze mit diesen Anionen bilden.
Versuch 27.1: Mineralien aufstellen
Die Chemie der Alkalielemente besteht im Wesentlichen daraus, das eine Valenzelektron abzugeben und Ionenverbindungen aufzubauen. Diese sind mit einer Vielzahl von Anionen wasserlöslich. Beispiele sind die Carbonate und die Hydroxide, die mit fast allen anderen kationbildenden Elementen schwerlöslich sind.
Die Elemente sind dementsprechend starke Reduktionsmittel, die leicht ihr Valenzelektron übertragen. Dabei entstehen nicht immer die erwarteten Produkte. Besonders ungewöhnlich ist die Umsetzung beim Verbrennen an der Luft, bei der Lithiumoxid, aber Natriumperoxid und Kalium-, Rubidium- und Caesiumhyperoxid entstehen:
2 Li + ½ O2 → Li2O
2 Na + O2 → Na2O2
M + O2 → MO2 (M = K, Rb, Cs)
Die Elemente sind typische Metalle, die allerdings durch ihre ungewöhnlich geringe Härte auffallen.
Versuch 27.2: Natrium als weiches Metall
Da auch die Hydroxide reichlich in Wasser löslich sind, gibt es keine passivierenden Schichten, so dass die Alkalimetalle entsprechend der Lage der elektrochemischen Potentiale auch mit Wasser heftig reagieren.
Versuch 27.4: Reaktion von Lithium, Natrium und Kalium mit Wasser
• M + H2O → MOH + ½ H2 (M = Li, Na, K)
Die hohe Löslichkeit von Alkalisalzen erlaubt es, die Regeln der Keimbildung und des Keimwachstums (siehe bei Schwefel) zu studieren. Im Versuch wird gezeigt, wie die gehemmte Keimbildung von Natriumsulfat aufgehoben werden kann.
Versuch 27.8: Kristallisation einer übersättigen Glaubersalzlösung
Während in der Natur nur edle Metalle gediegen vorkommen (Cu, Ag, Au, Pt), werden die meisten Gebrauchsmetalle aus Oxiden, Sulfiden oder Carbonaten gewonnen. Der eigentliche Rohstoff für den technischen Prozess der Metallgewinnung ist in der Regel das Oxid. Sulfide und Carbonate werden vor der Reduktion zum Metall in das Oxid überführt. Sulfide werden geröstet, das heißt durch Erhitzen mit Luftsauerstoff wird Sulfid zu Schwefeldioxid, SO2, oxidiert. Ein Beispiel ist das Rösten von Kupfer(I)-sulfid:
2 Cu2S + 3 O2 → 2 Cu2O + 2 SO2
Carbonate geben beim Erhitzen CO2 ab und bilden die entsprechenden Oxide (vergleiche das „Kalkbrennen“). Seltene Metalle sind oft mit häufigeren vergesellschaftet und fallen bei der Verarbeitung der entsprechenden Erze als Nebenprodukte an (Cadmium bei der Zinkgewinnung; Gold bei der Kupfergewinnung).
In den als Rohstoffe für die Metallgewinnung eingesetzten Erzen haben die Metalle eine positive Oxidationsstufe, falls sie nicht gediegen vorliegen. Ziel der Verfahren zur Metallherstellung ist daher die Reduktion der Metallionen; sie unterscheiden sich in der Art und Weise, wie den Metall-Kationen Elektronen zugeführt werden.
Unedle Elemente wie Natrium, Magnesium oder Aluminium sind durch ihre stark negativen Normalpotentiale charakterisiert (−2,71; −2,35 und −2,69 V). In wässrigen Lösungen von Salzen dieser Kationen werden an der Kathode daher H3O+-Ionen reduziert, entsprechend dem höheren Normalpotential des Wasserstoffs. Die elektrochemische Reduktion dieser Elemente gelingt erst in Abwesenheit von leichter reduzierbaren Ionen, also in der Schmelze eines entsprechenden Salzes.
So wird Natrium durch Schmelzflusselektrolyse von Steinsalz, NaCl, hergestellt:
2 NaCl → 2 Na + Cl2
Aluminium ist das mengenmäßig bedeutendste Metall, das durch Schmelzflusselektrolyse gewonnen wird. Aluminium ist zugleich das wichtigste Nichteisenmetall überhaupt; es wird in größerer Menge hergestellt als zum Beispiel Kupfer, Zink oder Blei. Ausgangsstoff für die Elektrolyse ist nach den oben beschrieben Verfahren gereinigte Al2O3. Anders als im Fall des NaCl ist eine elektrolysierbare Schmelze nicht durch einfaches Erhitzen zugänglich, da der hohe Schmelzpunkt von Aluminiumoxid von mehr als 2000 °C dies unmöglich macht. Elektrolysiert wird daher die Schmelze eines als Lösungsmittel für Aluminiumoxid dienenden Salzes, und zwar Natriumhexafluoridoaluminat, Na3AlF6 (Kryolith), einem kationreichen Salz mit dem komplexen [AlF6]3−-Ion. Die Abbildung zeigt einen Strukturausschnitt.
Ausschnitt aus der Kristallstruktur des Kryoliths, Na3AlF6. Farbcode: beige-braun Al, violett Na, türkis F.
Eine Lösung von ca. 10 % Al2O3 in Kryolith schmilzt bei ca. 960 °C, in der Praxis kann man den Schmelzpunkt des Gemisches durch Zusatz weiterer Salze (Lithiumfluorid, LiF) zur Schmelze weiter senken. Die Elektrodenreaktionen, bezogen auf eine Formeleinheit Al2O3, sind:
Kathode: 2 Al3+ + 6 e− → 2 Al
Anode: 3 O2− + 3 C → 3 CO + 6 e−
Die Formulierung der Ionen ist eine grobe Näherung, da sowohl die Aluminium- wie auch die Oxid-Ionen in Form von Fluoroaluminaten wie [AlF5O]4− vorliegen. Die verwendeten Elektroden bestehen aus Graphit; während die die Bodenauskleidung der Zelle bildende Kathode beständiger ist, wird die Anode durch den formal entstehenden Sauerstoff zu Kohlenmonoxid oxidiert. Bei den heute üblichen Betriebsbedingungen (4,5 bis 5 V; 50 bis 300 kA pro Anlage, die aus 100 bis 200 in Reihe geschalteter Einzelzellen besteht) ergibt sich als Rohstoff- und Energieeinsatz für die Herstellung von 1 kg Aluminium: 2 kg Al2O3, ca. 4 kg Bauxit, 0,5 kg Anodengraphit und 14 bis 18 kWh elektrische Energie.
Während sich unedle Metalle aus wässriger Lösung nicht abscheiden lassen, können Metalle, die gegenüber Wasserstoff hinreichend edel sind, aus den Lösungen ihrer Salze in Wasser gewonnen werden. Man beachte, dass die Überspannung von Wasserstoff an einem Metall erheblich sein kann; bereits Zink ist daher edel genug, um aus einer wässrigen Lösung anstelle von H2 abgeschieden zu werden. Die Raffination von Kupfer ist ein besonderes Beispiel hierfür, da die benötigte Kupfersalz-Lösung erst im Verlauf der Elektrolyse entsteht.
Wird zwischen zwei Kupfer-Elektroden, die in eine leitende Lösung eintauchen, eine Spannung angelegt, so finden die folgenden Reaktionen statt:
Anode: Cu → Cu2+ + 2 e−
Kathode: Cu2+ + 2 e− → Cu
Die notwendige Spannung ist klein, da keine Zellspannung kompensiert, sondern lediglich der Widerstand des Elektrolyten überwunden werden muss. Besteht die Anode aus Rohkupfer und die Kathode aus Reinkupfer, so findet eine Reinigung des Kupfers statt: (a) Unedle Verunreinigungen (Zink, Eisen) gehen mit in Lösung, scheiden sich aber bei der geringen Zellspannung nicht wieder ab, (b) edlere „Verunreinigungen“ (Silber, Gold, Platin) werden durch die nicht hinreichend aufgeladene Anode nicht oxidiert und sammeln sich im Anodenschlamm, aus dem sie gewonnen werden können. Die Reinkupferkathode wächst auf Kosten der Rohkupferanode. Die Zellspannung beträgt bei diesem Verfahren, nach dem auch Metalle wie Nickel, Silber und Gold gereinigt werden können, 0,3 V. Das gewonnene Kupfer hat eine Reinheit von 99,95 %.
Kohlenstoff ist in der Form von Koks das billigste Reduktionsmittel der Technik. Durch Umsetzen geeigneter Verbindungen mit Kohlenstoff werden die Metalle Cobalt, Nickel, Zinn, Blei, Wismut, Zink, vor allem aber Eisen hergestellt. Das Verfahrensprinzip besteht im einfachsten Falle in der Umsetzung des entsprechenden Metalloxids mit Kohlenstoff bei erhöhter Temperatur:
MOx + x C → M + x CO
Durch die Anwesenheit von Kohlenstoff wird der endothermen thermischen Zersetzung eines Metalloxids in Metall und Sauerstoff eine exotherme Komponente beigemischt, nämlich die Bildung von CO aus C und ½ O2. Auf diese Anpassung der thermodynamischen Randbedingungen kann lediglich bei den weniger stabilen Oxiden einiger Edelmetalle verzichtet werden.
Versuch 28.19: Thermische Zersetzung von Silber(I)-oxid
• Ag2O → 2 Ag + ½ O2
Dagegen benötigt das wichtigste Gebrauchsmetall, Eisen, im Hochofenprozess das Zusammenspiel mit der Kohlenstoff-Oxidation. Größe, und Form eines Hochofens und der Temperaturverlauf im Ofen sind in allen empfohlenen Lehrbüchern anschaulich dargestellt. Beachten Sie, dass neben dem Eisenerz und Koks „Zuschläge“ zugemischt werden, in denen die Regeln von Bjerrums Base-Antibase-Theorie angewendet werden. Je nach der Verunreinigung des Erzes (Gangart) so ausgewählt wird, dass Schlacke entsteht. Liegt eine silicium- und aluminiumreiche Gangart vor, dann wird Calciumoxid in Form von Kalkstein oder Dolomit zugefügt, ist die Gangart hingegen kalkreich, dann wird aluminiumhaltiges SiO2 (Feldspat) zugeschlagen. Die dann vor allem aus aluminiumhaltigem Calciumsilicat bestehende Schlacke wird als Baumaterial verwendet. Es wird dabei angestrebt, eine Schlacke mit einer ähnlichen Zusammensetzung wie bei Portlandzementklinker herzustellen („Hochofenzement“). Vergleichen Sie in den Lehrbüchern die Grundlagen der Stahlherstellung – auch dort wiederholen sich diese Prinzipien.
Wann lässt sich ein Metall durch Reduktion des Oxids mit Kohlenstoff gewinnen? Es muss erstens so edel sein, dass die reduzierende Wirkung von Kohlenstoff ausreicht, und zweitens darf das entstehende Metall mit unumgesetztem Kohlenstoff keine stabile Verbindung bilden. Letzteres ist jedoch ist für etliche Metalle der Fall. So bilden Titan und Wolfram die Carbide wie TiC, W2C oder WC.
Neben Aluminium (Thermitverfahren) werden Alkali- und Erdalkalimetalle als Reduktionsmittel zur Gewinnung von unedleren Metallen eingesetzt. So wird das als Stahlveredler verwendete Vanadium („Chrom-Vanadium-Stahl“) mit Hilfe von Calcium hergestellt:
V2O5 + 5 Ca → 2 V + 5 CaO
Titan wird mit Hilfe von mit Magnesium hergestellt. Ausgangsstoff ist das Chlorid, das aus dem Oxid, dem natürlich vorkommenden Titan-Rohstoff, bei 900 °C gewonnen wird gemäß
TiO2 + 2 C + 2 Cl2 → TiCl4 + 2 CO
Das Chlorid wird bei 800 °C unter Argon als Schutzgas mit Natrium oder Magnesium als Reduktionsmittel umgesetzt (Halogenmetallurgie):
TiCl4 + 2 Mg → Ti + 2 MgCl2
Titan entsteht bei diesem Verfahren als Schwamm, der dann unter Argon aufgeschmolzen wird. Titan ist wie Vanadin ein Stahlveredler (Titanstahl), aber auch Grundmetall für Titanlegierungen. Der besondere Wert von Titan besteht darin, dass es unter den Leichtmetallen (Dichte: 4,50 g cm−3, ein Schwermetall hat eine größere Dichte als 5 g cm−3) ungewöhnliche Eigenschaften aufweist. So hat es einen hohen Schmelzpunkt, große Festigkeit – auch bei hoher Temperatur – und hervorragende Korrosionsbeständigkeit.
Weitere Metalle, die halogenmetallurgisch hergestellt werden, sind Zirkon, die Lanthanoide und die Actinoide.
Die Flüchtigkeit ihrer Iodide ist die Grundlage einer Feinreinigungsmethode für die Elemente Titan, Zirkon, Hafnium und Vanadin. Das Verfahren beruht auf dem folgenden chemischen Gleichgewicht:
Ti + 2 I2 ⇄ TiI4 ΔrH° = −376 kJ mol−1
Die Gleichgewichtsreaktion ist exotherm in Bezug auf die Iodidbildung. Bei tiefer Temperatur ist daher die Bildung des flüchtigen (?) TiI4 bevorzugt, beim Erhitzen verschiebt sich das Gleichgewicht zugunsten der linken Seite.
Beim van-Arkel-de-Boer-Verfahren wird bei ca. 500 °C Rohtitan mit wenig Iod in einem abgeschlossenen Gefäß zu TiI4 umgesetzt. Im Gasraum befindet sich ein glühender Wolfram-Draht (T > 1100 °C), an dem sich Titan gemäß der Umkehrreaktion abscheidet. Das frei werdende Iod diffundiert durch den Gasraum zurück zum Rohtitan, reagiert erneut, usw. – es findet chemischer Transport statt.
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|
| Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn |
| Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd |
| La | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg |
| Mortimer: 29 Binnewies: 23 Riedel: 5.4 |
Für die Hauptquantenzahl n = 3 ergeben sich die Nebenquantenzahlen l = 0, 1 und 2 oder s-, p- und d-Orbitale. Von l = 2 leiten sich die fünf Orientierungsquantenzahlen ml = −2, −1, 0, 1, 2 ab entsprechend fünf d-Orbitalen. d-Orbitale sind durch zwei Knotenebenen charakterisiert, es handelt sich um Orbitale mit vier „Lappen“, die bei den drei Orbitalen mit den Kennungen xy, xz und yz zwischen die Achsen des cartesischen Referenzsystems weisen, während die vier Lappen des x2−y2-Orbitals auf den Koordinatenachsen liegen:
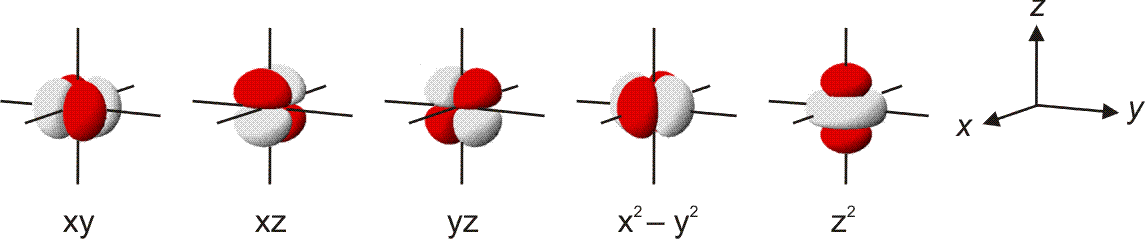
Das z2-Orbital fällt nur scheinbar aus dem Rahmen. Seine abweichende Gestalt ergibt sich aus dem Umstand, dass ebenso wie die drei Orbitale xy, xz und yz drei weitere Orbitale formuliert werden können: x2−y2, y2−z2 und z2−x2. Da sich aus den oben genannten Formeln nur fünf Orbitale ergeben, werden die beiden letzten Wellenfunktionen linearkombiniert (auch hier wieder ohne Normierung) zu: 2 z2 − x2 − y2 (in manchen Büchern auch 3 z2 − r2 mit r2 = x2 + y2 + z2); „z2“ ersetzt eine dieser beiden unhandlichen Bezeichnungen.
Das 3d-Niveau wird im Periodensystem in der ersten Übergangsreihe (Sc bis Zn) aufgefüllt, nachdem bei K und Ca bereits das 4s-Orbital besetzt wurde. Die für die Reihung im Periodensystem maßgebliche energetische Orbitalabfolge im Grundzustand der Atome – 4s stabiler als 3d – kehrt sich bei der Bildung von Verbindungen um. Bei der Diskussion von Verbindungen der Übergangsmetalle werden die vorhandenen Elektronen daher stets dem d-Niveau zugeordnet. Beispiel: Ein Fe-Atom hat im atomaren Grundzustand die Konfiguration 4s23d6, Eisen(II) aber nicht 4s23d4, sondern 3d6, Eisen(III) nicht 4s23d3, sondern 3d5.
Unter den Verbindungen der Übergangsmetalle kommt die oktaedrische Koordination ähnlich häufig vor wie die tetraedrische bei den Hauptgruppenelementen. Die chemische Bindung in Übergangsmetallverbindungen lässt sich daher besonders anschaulich für den Fall der oktaedrischen Umgebung ableiten.
Als Ausgangspunkt soll das Hexafluoridogallat(III)-Trianion, [GaF6]3−, dienen, das schwere Homologe des [AlF6]3−-Ions, dem Anion in Kryolith:
Eine Beschreibung der Bindungsverhältnisse könnte wie bei SF6 von 4e-3z-Bindungen ausgehen. Hinzu kommt nun, dass ein Gallium-Atom erheblich elektropositiver als ein Schwefelatom ist. Die Bindung ist also viel polarer. Bei der Formulierung von Lewis-Formeln gewinnt daher eine rein ionische Grenzformel (rechts) an Gewicht (E = Zentralatom in einer 4e-3z-Bindung, X = elektronegativer Substituent):
Wird eine rein ionische Formulierung als Ausgangspunkt gewählt, würde man von sechs Fluorid-Ionen und einem Ga3+-Ion ausgehen. Dieses hat die Elektronenkonfiguration [Ar]3d104s04p0. Im Sinne der 4e-3z-Betrachtung würden dann kovalente Anteile berücksichtigt, indem unter Beachtung der Oktett-Regel das 4s- und die drei 4p-Orbitale einbezogen würden. Die Aussage, dass bei Hauptgruppenelementen keine d-Orbitalbeteiligung bei scheinbar oktett-aufgeweiteten Molekülen oder Ionen vorliegt, betrifft die leeren 4d-Orbitale des Galliums. Man beachte also: keine d-Orbitalbeteiligung bei Verbindungen des Typs SF6 bedeutet, dass keine Erweiterung des Oktetts durch Bindung in leere d-Orbitale derselben Hauptquantenzahl erfolgt. (Als mögliche Ursache wird zum Beispiel diskutiert, dass diese d-Orbitale zu diffus sind und eine nur schlechte Überlappung zulassen.) Die vollbesetzten 3d-Orbitale des Ga3+-Ions werden wie alle vollbesetzten Orbitale unterhalb der Valenzschale nicht für Ga-F-Bindungen herangezogen.
Welche Wechselwirkungen zwischen d-Orbitalen und sechs auf das Zentralatom gerichteten Elektronenpaaren von sechs Fluorid-Ionen (der Einfachheit halber reine p-Orbitale) sind überhaupt denkbar? Wir betrachten zuerst die drei d-Orbitale der Ausrichtung xy, xz und yz: da die Orbitallappen zwischen die Achsen weisen, entlang derer die Fluor-Orbitale ausgerichtet sind, ist die Überlappung 0. Anders ist dies für die beiden Orbitale der Ausrichtung x2−y2 und z2. Beide Orbitale können im Prinzip mit Fluor-Orbitalen bindend und antibindend kombiniert werden. So ergibt sich für das d(z2)-Orbital (gezeigt ist die bindende Wechselwirkung):
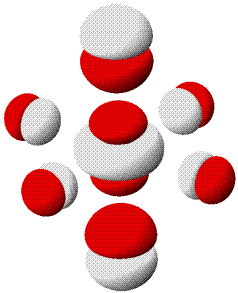
In ähnlicher Weise ergeben sich ausgehend vom d(x2−y2)-Orbital bindende und antibindende Wechselwirkungen in der xy-Ebene; gezeigt ist wieder die bindende Kombination:
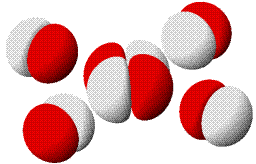
Zu berücksichtigen sind also zwei bindende und zwei antibindende Molekülorbitale. Wie tragen diese Wechselwirkungen zu den Ga-F-Bindungen im [GaF6]3−-Ion bei? – Nicht viel. Die Wechselwirkung von Fluorid mit den leeren 4d-Orbitalen des Galliums entspricht der mehrmals diskutierten Oktetterweiterung, die aufgrund quantitativer Berechnungen als wenig bedeutend eingestuft wird. Die Wechselwirkung zwischen Fluor-Orbitalen und dem 3d-Niveau führt natürlich erst recht zu keinem Beitrag zur Bindung, da sowohl die Metall- wie auch die Ligandorbitale mit jeweils einem Elektronenpaar gefüllt sind, wodurch sich gleich viele bindende wie antibindende Wechselwirkungen ergeben (man vergleiche den lehrbuchüblichen He2-Fall):
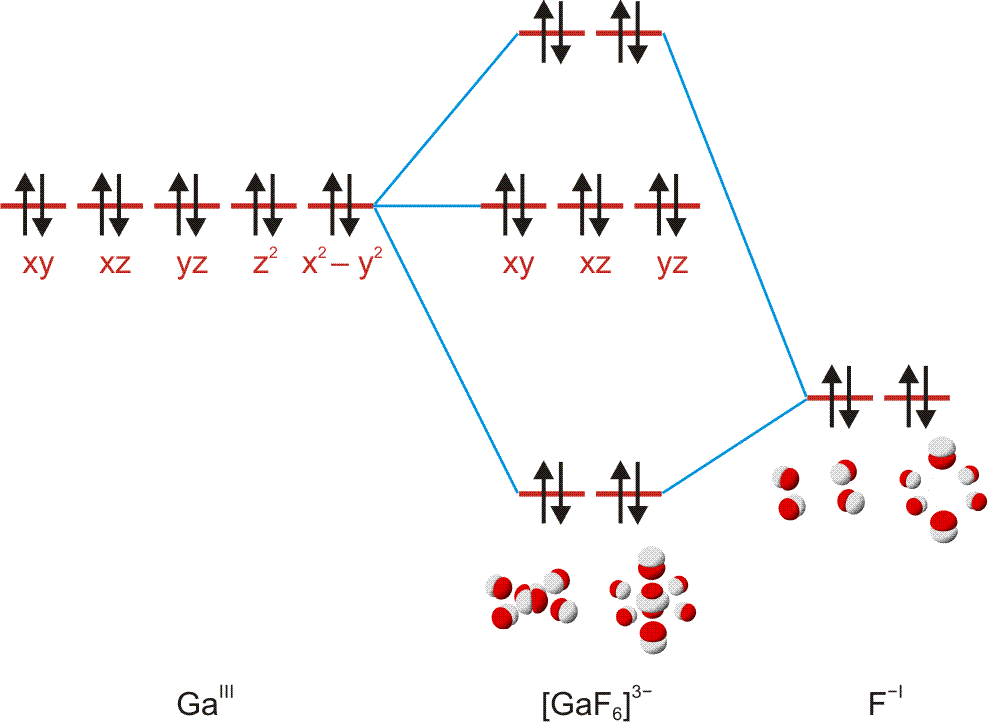
Auschnitt aus dem MO-Schema des [GaF6]3−-Ions. Bei Ga3+ sind die 3d-Orbitale berücksichtigt, bei Fluor sind diejenigen beiden der insgesamt sechs Orbitalkombinationen eingezeichnet, die jeweils eine bindende und antibindende Wechselwirkung mit dem d(z2)- und dem d(x2−y2)-Orbital des Galliums eingehen können (die vier übrigen Kombinationen wechselwirken mit dem 4s-Orbital und den 4p-Orbitalen so wie bei SF6 gezeigt). Es wurde davon ausgegangen, dass die Fluorid-Orbitale stabiler als die Gallium-Orbitale sind (in der Literatur wird dies das „normale Schema“ genannt), was jedoch ein kritischer Punkt ist.
Für das [GaF6]3−-Ion bleibt es nach dieser Betrachtung bei der Analogie zu SF6: acht Elektronen verteilen sich über sechs Bindungen, an denen das Gallium-Zentralatom mit seinen 4s- und 4p-Orbitalen beteiligt ist. Diese Situation ändert sich jedoch, wenn nun die im Periodensystem vor Gallium stehenden Elemente der ersten Übergangsreihe betrachtet werden. Ein stabiles Hexafluoridometallat-Ion in der dreiwertigen Stufe des Zentralatoms ist zum Beispiel bei Eisen bekannt.
Dieselbe Betrachtung wie beim Hexafluoridogallat(III)-Ion führt nun zu einem anderen Ergebnis. Als Folge der Elektronenkonfiguration [Ar]3d5 sind nämlich 5 Elektronen weniger als beim Gallat in das Schema eingefüllt. Da hierdurch weniger Elektronen in antibindenden Zuständen vorliegen, erhöht sich die Bindungsordnung. In der Summe kommt so zur Wechselwirkung der Fluorid-Elektronenpaare mit 4s- und 4p-Orbitalen ein Beitrag zur Bindung durch d-Orbitale vom Typ (n−1)d hinzu (man beachte: der Beitrag durch d-Orbitale der gleichen Hauptquantenzahl wie die Valenz-s- und Valenz-p-Niveaus ist weiterhin bedeutungslos):
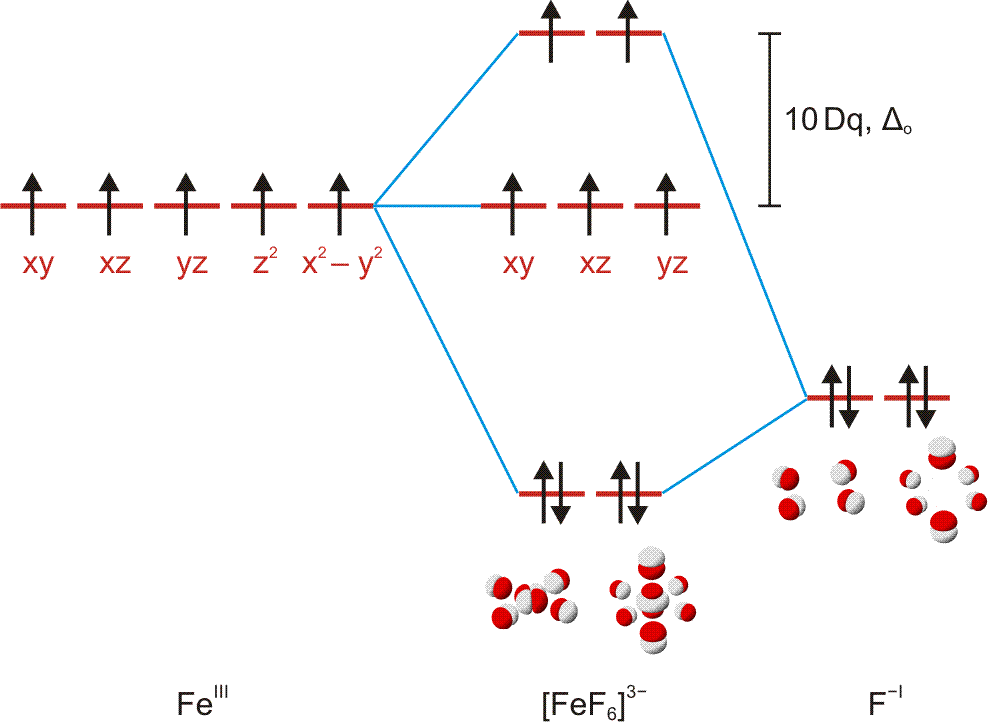
Weiter unten wird auf zwei Einzelheiten des Schemas eingegangen: (1) auf die Bedeutung des Energieunterschieds zwischen den nichtbindenden und den antibindenden Orbitalen – der mit 10 Dq oder Δo abgekürzten Ligandenfeldaufspaltung, und (2) auf die gewählte Elektronenverteilung, bei der die nichtbindenden und antibindenden Orbitale nicht nach dem Aufbauprinzip, sondern nach der Hundschen Regel gefüllt wurden; dies ist der high-spin-Fall.
Die Anionen Hexafluoridogallat(3−) und Hexafluoridoferrat(3−) sind „Metallkomplexe“, auch kurz „Komplexe“, sie sind die Anionen von Koordinationsverbindungen. Dies sind Verbindungen, in denen mindestens 1 koordinative Bindung vorkommt.
Die Bildung von Komplexen kann die Reaktivität von Metallverbindungen sehr weitgehend verändern. So beruht ein empfindlicher Eisen(III)-Nachweis auf der Bildung des intensiv roten Eisen(III)-thiocyanats. Die Zugabe von Fluorid-Ionen zu einer Eisen(III)-thiocyanat-Lösung entfärbt diese – offensichtlich wird durch die Bildung der [FeF6]3−-Ionen die Konzentration an „freien“ Eisen(III)-Ionen so weit erniedrigt, dass der charakteristische Nachweis ausbleibt.
Versuch 28.3: „Maskieren“ von Eisen(III)
• Fe(SCN)3 + 6 F− → [FeF6]3− + 3 SCN−
Der Komplex ist sehr stabil, er wird daher in der analytischen Chemie zum „Maskieren“ von Eisen(III) benutzt. „Stabil“ bezieht sich bei einem komplexen Ion auf das Bildungsgleichgewicht des Komplexes aus dem Zentralmetall(-Ion) und den Liganden. Der Komplex ist umso stabiler, je weiter das Gleichgewicht auf der rechten Seite liegt, je größer also die Gleichgewichtskonstante ist; für das Hexafluoridoferrat(III)-Ion gilt:
Fe3+ + 6 F− ⇄ [FeF6]3−
und
Man beachte einen ärgerlichen Umstand: eckige Klammern sind zum einen gebräuchlich, um einen Komplex zu kennzeichnen, zum anderen aber bedeuten sie „Konzentration als Vielfache der Standardkonzentration 1 mol L−1“; es ist eben so. Wird die Komplexbildung nicht schrittweise beschrieben, indem für die Anlagerung eines jeden einzelnen Fluorido-Liganden an das Zentralmetall ein Gleichgewicht formuliert wird, sondern wird – wie gezeigt – die Gesamtbilanz betrachtet, so heißt die Gleichgewichtskonstante Bruttokomplexbildungkonstante; sie wird üblicherweise mit β abgekürzt. Ein Index wie hier „16“ wird meistens angegeben, er bedeutet, dass der Komplex aus 1 Zentralatom und 6 Liganden besteht.
Was aber bedeutet Fe3+? Gemeint ist hier wie auch in ähnlichen Formulierungen, die sich aus dem Massenwirkungsgesetz ableiten (Löslichkeitsprodukt, Nernstsche Gleichung, etc.), nicht etwa das „nackte“ Eisen(III)-Ion, sondern ein Eisen(III)-Ion im Standardzustand. Dieser ist definiert durch eine wässrige Lösung, in der das Eisen-Ion idealerweise mit nichts anderem wechselwirkt als mit Wassermolekülen. Unter diesen Umständen liegt das Ion hydratisiert vor, und zwar als Hexaaquaeisen(III)-Ion – also auch einem Komplex, jetzt mit Wassermolekülen als Liganden.
Hydratisierte Ionen dreiwertiger Metalle wie Ti3+, V3+, Cr3+, Fe3+, Co3+, Al3+ und Ga3+ zeigen typische Eigenschaften von Metallkomplexen. Um sie in Lösung zu untersuchen, sind stark saure Bedingungen notwendig, um die Bildung von Hydroxido-Komplexen (und letztlich die Bildung der Metallhydroxide) zu unterbinden. Um die Farbe der Komplexe zu messen, wird daher typischerweise das Perchlorat des dreiwertigen Metalls in starker Perchlorsäure gelöst. In dieser Lösung kann von [MIII(H2O)6]3+-Ionen ausgegangen werden. Strukturparameter, aber auch magnetische und optische Eigenschaften können an kristallinen Proben untersucht werden, da die Hexaaquakomplexe dreiwertiger Metalle zur Bildung von Alaunen neigen. Alaun ist KAl(SO4)2·12 H2O, isotyp kristallisieren zahlreiche Verbindungen mit verschiedenen einwertigen Ionen außer Kalium und eben zahlreichen dreiwertigen Metall-Ionen. Die Formel der Alaune wird besser als MI[MIII(H2O)6](SO4)2·6 H2O angegeben, aus der die Zuordnung von 6 Wassermolekülen zu einem dreiwertigen Zentralatom hervorgeht. Die Abbildung zeigt die Struktur des Hexaaquachrom(III)-Ions im violetten, in großen Oktaedern aus wässriger Lösung kristallisierenden Kaliumchromalaun, KCr(SO4)2·12 H2O:
Eine erste charakteristische Eigenschaft der Übergangsmetallvertreter der Alaune ist ihre Farbe, die stets sehr blass ist verglichen mit stark absorbierenden Chromophoren wie dem Permanaganat-Ion. Neben dem Hexaaquachrom(III)-Ion ist das Hexaaquatitan(III)-Ion, [TiIII(H2O)6]3+ ist in dieser Hinsicht typisch. Es entsteht als blassviolettes Ion bei der Reduktion saurer TiIV-Lösungen mit Zink
Versuch 28.2: Herstellung von violettem Hexaaquatitan(III)
• 2 TiIVO2+ + Zn + 2 H3O+ + 9 H2O → 2 [TiIII(H2O)6]3+
Die blass-violette Farbe des Hexaaquatitan(III)-Ions lässt sich aus der oben für den Hexafluoridoferrat(III)-Komplex angestellten MO-Betrachtung ableiten. Das d1-Ion [TiIII(H2O)6]3+ bietet die nur mit einem Elektron besetzte Orbitalgruppe d(xy), d(xz) und d(yz) als Ziel von Elektronenanregungen an, die von vollbesetzten stabileren Orbitalen ausgehen. Das gleiche gilt für die beiden hier leeren antibindenden Orbitale im gezeigten Schema, die darüberhinaus auch das eine d-Elektron nach dessen Anregung aufnehmen können. Ein Vergleich mit den stets farblosen Ionen ähnlichen Aufbaus, die anstelle einer dn-Konfiguration eine d0- oder d10-Anordnung aufweisen, zeigt, dass die Farbe von Übergangsmetallkomplexen auf einer Anregung von d-Elektronen beruht. Im Fall des Hexaaquatitan(III)-Ions also der Anregung, welche die Ligandenfeldaufspaltung überwindet.
Solange das MO-Schema die oben gezeigte Orbitalabfolge aufweist („normales Schema“), lassen sich zahlreiche Eigenschaften von Komplexen einschließlich der Farben durch ein verblüffend einfaches Modell beschreiben, dem Kristallfeldmodell. Diese sehr alte Modellvorstellung wurde zur Beantwortung der allgemeinen Frage entwickelt, ob und wie sich Orbitalenergien ändern, wenn ein Atom zum Beispiel in einem NaCl-Typ-Kristall in oktaedrischer Umgebung auf dem Platz eines der Ionen sitzt. Für die d-Orbitale eines Kations, das oktaedrisch von sechs Anionen umgeben ist, ergibt eine solche Betrachtung, dass die beiden Orbitale d(x2−y2) und d(z2) besonders stark destabilisiert sein sollten, da sie den negativen Ladungen der Anionen besonders nahe kommen und so die größte Abstoßung erfahren. Auch die übrigen drei Orbitale erfahren eine Abstoßung, diese ist jedoch geringer, da die Lappen der d(xy)-, d(xz)- und d(yz)-Orbitale zwischen die negativen Ligandladungen weisen und nicht direkt darauf. Der Energieunterschied ist die erwähnte Ligandenfeldaufspaltung, die beim Gebrauch des Kristallfeldmodells meist Kristallfeldaufspaltung genannt wird. Es werden die erwähnten Symbole 10 Dq oder Δo verwendet. Die Anregung des d-Elektrons durch die Absorption von Licht stellt sich im Kristallfeldmodell wie folgt dar:
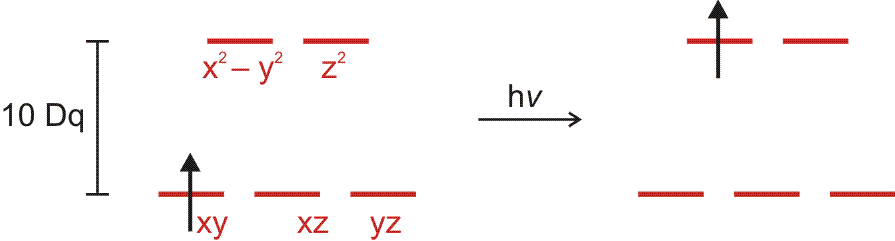
Das Modell ist so einfach wie es ist, da die Liganden eines Komplexes nicht mit ihren Orbitalen betrachtet werden. Orbitale werden nur am Zentralmetall zugelassen, während die Liganden zu negativen Punktladungen vereinfacht werden. Ungewohnt ist, irgendwelche Beobachtungen mit einem Abstoßungsmodell zu beschreiben. Bislang wurde die Annäherung zweier Atome nicht im Sinne einer Abstoßung interpretiert, sondern als Voraussetzung für eine Bindung. Wieso lässt sich also mit dem Kristallfeldmodell irgend etwas erklären? Das oben diskutierte MO-Schema beantwortet diese Frage. Die vom Kristallfeldmodell allein betrachteten Metallorbitale sind die Grenzorbitale des Komplexes (bei normalem Schema). In diesem Grenzorbitalbereich finden sich nichtbindende und antibindende Wechselwirkung, deren Verstärkung in der Tat mit einer erhöhten Abstoßung zwischen Metall und Ligand einhergeht. Das Kristallfeldmodell modelliert nun diesen antibindenden Grenzorbitalbereich und kann zutreffende Aussagen über die Elektronenstruktur in diesem für die Eigenschaften maßgeblichen Bereich machen – solange man eine Frage nicht stellt, für die besser zum Beispiel ein MO-Schema verwendet wird: was hält den Komplex eigentlich zusammen?
Hält man sich diese Einschränkungen vor Augen, lassen sich mit dem Kristallfeldmodell viele nützliche Aussagen ableiten, zum Beispiel …
Eine besonders charakteristische Reaktionen eines Metalls ist die Bildung von Komplexen mit Ammoniak-Molekülen als Liganden („Ammin-Komplexe“). So bilden Kupfer(II)-Salzlösungen bei der Zugabe von überschüssigem Ammoniak stabile tintenblaue Komplexe, deren Entstehung den analytischen Nachweis von Kupfer(II) leicht macht. Die Bildungstendenz der Komplexe, vor allem des Tetraammin-kupfer(II)-Ions, ist hoch. Es bildet sich auch ausgehend vom farblosen Tetrammin-kupfer(I)-Ion, einem d10-Komplex, bei dessen Oxidation mit Luftsauerstoff.
Versuch 28.4: Ammin-kupfer(II)-Komplexe
• [Cu(H2O)6]2+ + 4 NH3 → [Cu(NH3)4(H2O)2]2+ + 4 H2O
Versuch 28.15: Oxidation des farblosen Ammin-kupfer(I)-Komplexes
• 4 [Cu(NH3)4]+ + O2 + 10 H2O → 4 [Cu(NH3)4(H2O)2]2+ + 4 OH−
Die Struktur des Kupfer(II)-Komplexes in Kristallen eines Carbonsäuresalzes ist erstaunlich:
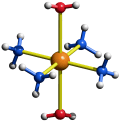
Die oktaedrische Gestalt des Komplexkations ist zwar deutlich zu sehen, sie ist jedoch merklich verzerrt: während die Cu-N-Bindungen nur 203 pm lang sind, sind die beiden Cu-O-Abstände 251 pm lang. Der Verdacht liegt natürlich nahe, dass zwischen Aqua- und Ammin-Ligand doch ein größerer Unterschied besteht als vermutet. Eine ähnliche Beobachtung wird jedoch beim homoleptischen Aquakomplex [Cu(H2O)6]2+ gemacht (ein homoleptischer Komplex hat nur eine Ligandsorte). Das folgende Bild zeigt die Struktur des Hexaaquakupfer(II)-Ions im Perchlorat [Cu(H2O)6](ClO4)2, bei dem die Sauerstoffatome der vier Aqua-Liganden in der Äquatorebene 195 pm vom Kupferatom entfernt sind, die axialen O-Atome jedoch 238 pm:
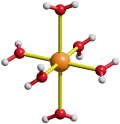
Ein Blick auf die Elektronenkonfigurationen des Zentralmetalls zeigt die Ursache der Verzerrung. Kupfer(II) hat [Ar]3d9-Konfiguration. Für eine regulär oktaedrische Umgebung zeigt das Kristallfeldmodell zwei energiegleiche Konfigurationen, die sich nur hinsichtlich der Besetzung der unstabilsten Orbitale unterscheiden (x und y in der Ebene der kürzeren Cu-O-Kontakte, z entlang der längeren): (x2−y2)2(z2)1 und (x2−y2)1(z2)2. Wir greifen nun von diesen beiden „entarteten“ Möglichkeiten die Besetzung (x2−y2)1(z2)2 heraus. Da nun ein Orbital doppelt besetzt ist, das andere aber nur einfach, ist die Abstoßung durch die Liganden unterschiedlich. Eine Minimierung der Abstoßungsenergien ist nun möglich, indem die Liganden vom doppelt besetzten z2-Orbital wegrücken und zum Ausgleich die Liganden in der xy-Ebene sich etwas auf das nur einfach besetzte Orbital zubewegen. Genau das zeigt die Strukturanalyse, der Komplex ist Jahn-Teller-verzerrt. Beachten Sie, dass eine Betrachtung, die von der (x2−y2)2(z2)1-Konfiguration ausgegangen wäre, anstelle eines über eine Achse gestreckten Oktaeders ein gestauchtes Oktaeder ergeben hätte. In diesem Sinne sagt das Jahn-Teller-Theorem voraus, dass eine Verzerrung erwartet werden darf, um die Struktur an eine ungleiche Elektronenverteilung anzupassen, aber nicht, in welcher Richtung die Verzerrung realisiert ist.
Wie kann nun erkannt werden, ob eine Jahn-Teller-Verzerrung zu erwarten ist? Komplexe und andere molekulare Spezies erleiden immer dann eine Jahn-Teller-Verzerrung, wenn sie in der hohen Symmetrie mehr als eine energiegleiche Elektronenverteilung zulassen. Benutzen Sie also das Kristallfeldmodell, um die Orbitalaufspaltung bei hoher Symmetrie darzustellen. Gibt es nun mehr als eine energiegleiche Möglichkeit, die d-Elektronen in das Schema einzufüllen, ist eine Verzerrung zu erwarten. Welche oktaedrischen Ionen sind also keine Jahn-Teller-Ionen?
Lösungen, die das Hexaaquacobalt(II)-Ion enthalten, haben eine blass-rosa Farbe. Das Chromophor, das [Co(H2O)6]2+-Ion, hat leicht verzerrt oktaedrische Struktur (hier im Sulfat; man beachte, dass die H-Atom-Lagen offensichtlich nicht sehr präzise bestimmt wurden):
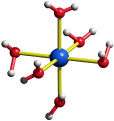
Auch hier ist das Oktaeder gestreckt (4 × 207 pm, 2 × 211 pm), aber viel schwächer als im analogen Kupfer-Ion – eine Beobachtung, die meist gemacht wird, wenn die Entartung in den Orbitalen des Typs d(xy) auftritt. Die Farbe der Lösung ändert sich sehr auffallend, vor allem in ihrer Intensität, wenn Salzsäure zugegeben wird.
Versuch 28.5: Reaktion von Salzsäure mit Cobalt(II)-chlorid-Lösung
• [Co(H2O)6]2+ + 4 Cl− → [CoCl4]2− + 6 H2O
Die Intensität der Lichtabsorption der gebildeten Cobalt-Spezies liegt nun in der gleichen Größenordnung wie bei Permanganat und organischen Farbstoffen. Zugleich ist der Aufbau des gebildeten Cobaltkomplexes unerwartet. Das Bild zeigt die Struktur des komplexen Anions in Kristallen von Cs3[CoCl4]Cl:
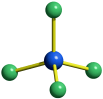
Es ist ein tetraedrischer Komplex entstanden. Das Kristallfeldmodell lässt sich auch hier anwenden. Im Gegensatz zum Oktaeder gibt es nun nicht den Fall, dass eine Orbitalgruppe genau auf die Liganden zuweist und die andere ihnen maximal aus dem Weg geht. Als Folge ist die Kristallfeldaufspaltung kleiner und zwar nur noch ungefähr halb so groß wie bei einem oktaedrischen Komplex mit der gleichen Zentralmetall-Ligand-Kombination. Außerdem erfahren nun die Elektronen in den d(xy)-, d(xz)- und d(yz)-Orbitalen die größere Abstoßung.
Für die beiden abgebildeten Cobaltkomplexe ergeben sich so zwei unterschiedliche Ausspaltungsschemata. Man beachte die Elektronenkonfiguration d7 des Cobalt(II)-Zentralmetalls.
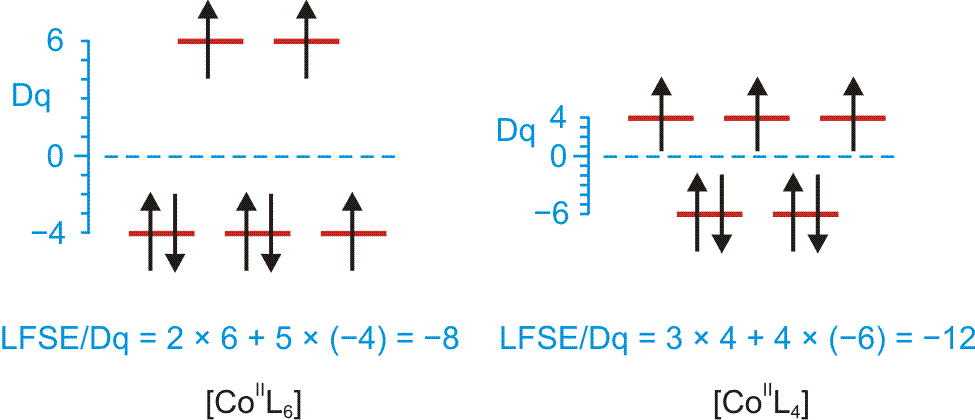
Im Schema ist eine Größe angegeben, die als Maß für den stabilisierenden Beitrag benutzt wird, der sich ergibt, wenn möglich viele unstabile Orbitale unbesetzt und möglichst viele stabile Orbitale besetzt sind – die Ligandenfeldstabilisierungsenergie, meist LFSE abgekürzt. Sie wird in Vielfachen von Dq oder Δ angegeben. Zu ihrer Berechnung wird die Kristallfeldaufspaltung auf einen Nullpunkt bezogen, der bei gleichmäßiger Besetzung zu einer LFSE von 0 führt. Die LFSE ergibt sich allgemein als Summe der Elektronenenergien, bezogen auf diese Skala. Man beachte, dass eine Energieangabe in Dq keine absolute Größe wie kJ oder eV ist. Da 10 Dq bei einem oktaedrischen Komplex ungefähr doppelt so groß wie bei einem tetraedrischen Komplex ist, sind die im Schema angegebenen Ligandfeldstabilisierungsenergien auf einer absoluten Skala näherungsweise gleich. Dies ist eine Eigenheit des high-spin-d7-Falls. Sie wird als Ursache dafür angesehen, dass Cobalt(II)-Komplexe ähnlich oft von der oktaedrischen Koordination abweichen wie es sonst nur für LFSE = 0 gefunden wird.
Warum ist das [CoCl4]2− viel intensiver farbig als die anderen Metallkomplexe, die bisher behandelt wurden? Der Unterschied wird deutlich, wenn die spektroskopischen Auswahlregeln beachtet werden. Eine elektronische Anregung findet dann ohne Einschränkungen statt, wenn zwei Regeln erfüllt sind. Die erste Regel verlangt, dass bei der Anregung der Spin des Elektrons erhalten bleibt. Das Schema zeigt, dass dies problemlos in beiden Fällen möglich ist: wird der häufigere Spin als α-Spin und der weniger häufige als β-Spin bezeichnet, so kann in beiden Fällen ein β-Spin von einem stabilen in ein unstabiles Orbital angeregt werden, ohne irgend eine Regel zu verletzen.
Die zweite Auswahregel, das Laporte-Verbot, ist etwas vertrackter. Eine übliche Formulierung geht so: eine Anregung ist erlaubt, wenn der Komplex kein Symmetriezentrum aufweist. Besitzt er ein Symmetriezentrum, müssen sich die beiden Orbitale, zwischen denen das Elektron verschoben wird, in ihrer Nebenquantenzahl um ±1 unterscheiden (d zu d verboten, p zu d erlaubt, etc.). Pro verletzter Regel nimmt die Farbintensität in grober Näherung um einen Faktor 100–1000 ab. Beachten Sie, dass für oktaedrische Komplexe alle Anregungen innerhalb der d-Orbitale Laporte-verboten und für alle tetraedrischen Komplexe Laporte-erlaubt sind. Im Rahmen dieser Vorlesung wird nicht darauf eingegangen, warum verbotene Übergänge überhaupt auftreten, wenn auch nur mit 1/1000–1/100 Intensität.
Das Hexafluoridoferrat(III)-Ion ist farblos. Wir diskutieren, warum. Dabei werden wir erkennen, dass Eisen(III)-chlorid auch farblos sein müsste, es ist jedoch intensiv gelb-orange. Die Ursache zeigt sich bei einer näheren Betrachtung des intensiv violetten Permanganat-Ions.
Permanganat-Lösungen, die das Anion MnO4− enthalten, sind ungefähr 500-mal intensiver violett als gleich konzentrierte Hexaaquatitan(III)-Lösungen, die Anregung ist also im Gegensatz zu der Laporte-verbotenen Anregung im Titan-Ion erlaubt. Da MnVII d0-Komplexe bildet, kommt als Anregung nur die Energieaufnahme durch ein Elektron vom Liganden in Frage. Bei Permanganat würde ein Oxido-Ligand für die Dauer der Anregung um ein Elektron oxidiert und das Mangan-Atom um ein Elektron reduziert werden – also zur Stufe +VI. Für die Dauer der Anregung findet eine Ladungsübertragung statt, engl. charge transfer. Der Begriff charge-transfer-Anregung, kurz CT-Anregung, hat sich auch im Deutschen eingebürgert. Der Ladungsübergang vom Ligand auf das Metall ist nicht der einzige mögliche, aber doch der häufigste (in älteren Büchern finden Sie den anschaulichen Begriff „Reduktionsbande“). Beachten Sie die einzelnen Faktoren, die beim Permanganat zusammenspielen: ein Ligand hoher Ladungsdichte trifft auf ein Zentralmetall mit einer hohen Ladung. Bei Eisenchlorid ist das Metall nicht so hoch geladen, dafür ist der Ligand polarisierbarer und die Energie der Anregung ist bei einem gelben Stoff höher.
Wir diskutieren die Farben und Reaktionen von Kupfer(II) mit den verschiedenen Halogenid-Ionen.
Versuch 28.14: Reaktion von Kupfer(II) mit Iodid
• 2 [Cu(H2O)6]2+ + 4 I− → 2 CuI + I2 + 12 H2O
Der Hexafluoridoferrat(III)-Komplex ist zwar außerordentlich stabil, die Kristallfeldaufspaltung, die durch die Fluorido-Liganden verursacht wird, ist jedoch an der unteren Grenze für Aufspaltungen bei Eisen(III)-Komplexen. In einer „Hitliste“ der von Liganden bei gleichbleibendem Zentralmetall erzeugten Feldaufspaltungen – der spektrochemischen Reihe – stehen Halogenido-, aber auch Oxido- oder Thiolato-Liganden auf sehr bescheidenen Plätzen. Einer der Spitzenreiter ist das Cyanid-Ion. Die mit ihm gebildeten Cyanido-Komplexe sind durch hohe Kristallfeldaufspaltungen charakterisiert. Auch mit Eisen(III) ist ein solcher Komplex bekannt, das Hexacyanidoferrat(III), das Anion des „roten Blutlaugensalzes“. Das folgende Bild zeigt den Aufbau in Kristallen von K3[Fe(CN)6]:
Die Ursache für die Stellung eines Liganden in der spektrochemsichen Reihe wird in dieser Vorlesung nicht behandelt, es soll jedoch die Folge einer hohen Feldaufspaltung gezeigt werden. Sowohl im Hexacyanidoferrat(III) als auch in dem durch Reduktion entstehenden, ungewöhnlich stabilen Hexacyanidoferrat(II), dem Anion des „gelben Blutlaugensalzes“, ist die Feldaufspaltung so groß, dass die Spinpaarungsenergie überschritten wird. Beide Salze sind low-spin-Komplexe, deren d-Elektronenverteilung dem Aufbauprinzip und nicht der Hundschen Regel folgt:
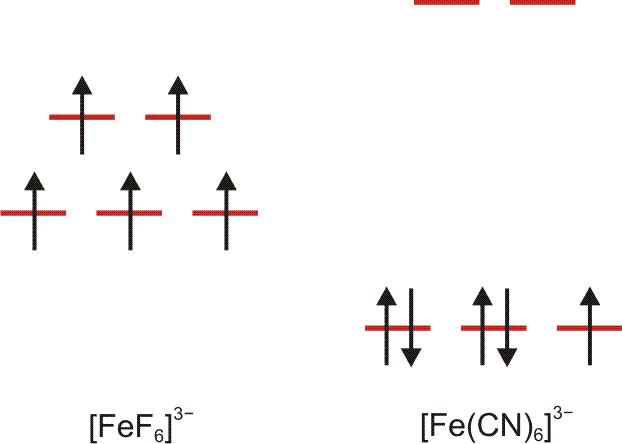
Die chemischen Eigenschaften werden vom Spinzustand des Zentralatoms geprägt. Das halbbesetzte d-Niveau im high-spin-d5-Ion geht mit einer besonderen Stabilität der dreiwertigen Stufe bei high-spin-Eisen-Verbindungen einher. Der low-spin-d5-Cyanokomplex ist dagegen ein brauchbares Oxidationsmittel, das leicht in die besonders stabile low-spin-d6-Konfiguration übergeht.
Liegen die Bedingungen für low-spin-Komplexe vor, so gewinnt bei der Konfiguration d8 die quadratisch-planare Anordnung von vier Liganden an Bedeutung. Das Kristallfeldmodell erlaubt auch in diesem Fall eine erste Analyse. Wir gehen kurz auf die Feldaufspaltung im quadratisch-planaren Fall ein.
Die quadratisch-planare Umgebung von low-spin-d8-Zentren ist nicht auf Komplexverbindungen wie [Ni(CN)4]2−, [Pt(NH3)2Cl2] („Cisplatin“) oder [AuCl4]− beschränkt, sondern sie bestimmt auch die Festkörperstrukturen einfach zusammengesetzter Verbindungen wie PdS:
Die Kristallstruktur von Palladium(II)-sulfid. Farbcode: türkis Pd, gelb S.
Die bisher besprochenen Liganden haben alle eine Gemeinsamkeit: sie sind sowohl einzähnige als auch terminale Liganden. Einzähnig bedeutet, dass der Ligand mit genau einem Atom an das Zentralmetall bindet, terminal bedeutet, dass der Ligand nicht zwei oder mehr Zentralmetallatome miteinander verbrückt.
Mehrzähnige Liganden bilden eine besondes wichtige Ligandengruppe. Bindet ein Ligandenmolekül mit mehr als einem Atom (dieses wird dann oft „Ligatoratom“ genannt) an ein Zentralatom, entsteht ein Chelatkomplex oder kurz Chelat. Die vom griechischen Wort für „Krebsschere“ abgeleitete Bezeichnung deutet auf die strukturelle Besonderheit, dass der Ligand zusammen mit den Zentralmetall einen „Chelatring“ ausbildet. Typische Chelatliganden sind Ethan-1,2-diamin („Ethylendiamin“, als Ligand abgekürzt: en) und die Dianionen der verschiedenen Weinsäure-Isomere (als di-anionischer Ligand abgekürzt: d-tart, l-tart, meso-tart, von „Tartrat“, dem Salz der Weinsäure). In der folgenden Struktur, die zwei Palladium(II)-Atome als Zentralatome enthält und daher ein zweikerniger Komplex ist, sind insgesamt vier Chelatringe zu sehen (beachten Sie wieder: PdII hat d8-Konfiguration, beide Pd-Atome sind dementsprechend quadratisch planar koordiniert). Außerdem tritt der Tartrato-Ligand nicht nur zweifach (an den COO−-Gruppen), sondern vierfach (an allen OH-Gruppen) deprotoniert auf (im Bild: Pd türkis, N blau, O rot, C und H grau):
Brückenliganden treten sehr häufig auf – immer dann, wenn Ligandmangel herrscht. Dies ist zum Beispiel dann der Fall, wenn ein Zentralmetall Hydroxidokomplexe bildet, der pH-Wert aber eher niedrig ist. Die weiter oben betrachteten Hexaaquakomplexe dreiwertiger Metalle sind typische Beispiele. Einige, das [Cr(H2O)6]3+-Ion zum Beispiel, reagieren mit Hydroxid-Ionen zum Metallhydroxid, das jedoch aufgrund der hohen Lewis-Acidität mancher dreiwertiger Metall-Ionen amphoter ist und sich in alkalischer Lösung unter Bildung von Hydroxido-Komplexen auflöst. So entsteht bei Chrom(III) ausgehend vom hydratisierten Metall-Ion über die Stufe des Chrom(III)-hydroxids hinaus das Hexahydroxido-chromat(III), dessen Struktur in Kristallen des tannengrünen Natriumsalzes einkernig und oktaedrisch ist:
Ein Einschub für Lehramtsstudierende: es gehört zu den zu vermittelnden Weisheiten, dass Modellvorstellungen ihre Grenzen haben. Hier ist eine besonders leicht vorführbare, bei der ein Schuss Natronlauge das Kristallfeldmodell kippt und das MO-Modell notwendig macht: Bei einem violetten Aquakomplex (der die Komplementärfarbe gelb absorbiert) führt der Basezusatz zur Entfernung von jeweils einem Proton pro Ligand. Hierdurch wird der Dipolligand Wasser zum anionischen Hydroxido-Ligand. Die Erhöhung der negativen Ladung an jedem Ligand führt zu einem grünen Komplex (der die Komplementärfarbe rot absorbiert). Rotes Licht transportiert weniger Energie als gelbes, 10 Dq verringert sich also bei einer Reaktion, welche die Liganden negativ auflädt. Was hätte man nach dem Kristallfeldmodell erwarten dürfen, das die Feldaufspaltung aus der Abstoßung von d-Elektron- und Ligandladung erklärt?
Weit vor der Hydroxidfällung, bei pH-Werten um vielleicht 3 beginnt die Bildung von Hydroxido-Komplexen bereits, Hydroxido-Liganden sind jedoch bei diesem pH-Wert rar. Dies sind die typischen Bedingungen, unter denen die Lewis-Basizität eines Liganden unter mehreren Lewis-aciden Metall-Ionen aufgeteilt wird. Bei Chrom(III) konnten solche Spezies in kristalliner Form isoliert werden. Das Bild zeigt ein [(H2O)4Cr(μ-OH)2Cr(H2O)4]4+-Kation in kristalliner Umgebung. Man beachte das für dreiwertige Metalle typische Verhalten gegenüber O-Liganden, kantenverknüpfte Oktaeder zu bilden (vgl. die Korund-Struktur). Beachten sie auch die Schreibweise für einen Brückenliganden (z.B. μ4 wenn er 4 Metallatome verbrückt, μ3 bei 3, μ2 bei 2; es gilt vereinfachend: μ2 = μ).
Die Stabilität eines Komplexes ist kaum damit korreliert, ob er Brückenliganden enthält oder nicht. Chelatliganden dagegen führen zu erheblich stabileren Komplexen als verwandte einzähnige Liganden. Dass die Beständigkeitskonstante typischerweise um ca. 10 Größenordnungen wächst, wenn einzähnige Liganden wie Ammoniak paarweise durch Chelatliganden wie Ethylendiamin ersetzt werden, ist der Chelateffekt. Er ist im wesentlichen entropisch verursacht. Wird als Ausgangspunkt ein Komplex mit sechs einzähnigen Liganden gewählt, so führt die Umsetzung mit drei zweizähnigen Chelatliganden zur Freisetzung von sechs Teilchen, also zu einem deutlichen Entropiegewinn.
Was „weiß“ die Natur von all diesen scheinbar abstrakten Regeln, wenn sie zum Beispiel Eisenzentren in Hämoglobin und Myoglobin zum Sauerstofftransport einsetzt? Offensichtlich alles: Der nichtkovalent in Globin eingelagerte Cofaktor Häm b bindet Eisen als Chelatkomplex. Der Spinzustand ist high-spin, bei Sauerstoffbindung schaltet er auf low-spin. Hämoglobin und Myoglobin sind molekulare Schalter. Wir sehen uns den Aufbau an.
1 Hartree = 627,5095 kcal mol−1 = 2625,500 kJ mol−1 = 27,2114 eV
1 cal = 4,184 J
1 eV × NA = 96,4853 kJ mol−1
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | He | ||||||||||||||||
| Li | Be | B | C | N | O | F | Ne | ||||||||||
| Na | Mg | Al | Si | P | S | Cl | Ar | ||||||||||
| K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr |
| Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe |
| Cs | Ba | La | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn |
| Aluminium | Al | 13 | 26,981538 |
| Argon | Ar | 18 | 39,948 |
| Arsen | As | 33 | 74,92160 |
| Barium | Ba | 56 | 137,327 |
| Beryllium | Be | 4 | 9,012182 |
| Bismut | Bi | 83 | 208,98038 |
| Blei | Pb | 82 | 207,2 |
| Bor | B | 5 | 10,811 |
| Brom | Br | 35 | 79,904 |
| Caesium | Cs | 55 | 132,90545 |
| Calcium | Ca | 20 | 40,078 |
| Chlor | Cl | 17 | 35,4527 |
| Fluor | F | 9 | 18,9984032 |
| Gallium | Ga | 31 | 69,723 |
| Germanium | Ge | 32 | 72,61 |
| Helium | He | 2 | 4,002602 |
| Indium | In | 49 | 114,818 |
| Iod | I | 53 | 126,90447 |
| Kalium | K | 19 | 39,0983 |
| Kohlenstoff | C | 6 | 12,0107 |
| Krypton | Kr | 36 | 83,80 |
| Lithium | Li | 3 | 6,941 |
| Magnesium | Mg | 12 | 24,3050 |
| Natrium | Na | 11 | 22,989770 |
| Neon | Ne | 10 | 20,1797 |
| Phoshor | P | 15 | 30,973762 |
| Rubidium | Rb | 37 | 85,4678 |
| Sauerstoff | O | 8 | 15,9994 |
| Schwefel | S | 16 | 32,066 |
| Selen | Se | 34 | 78,96 |
| Silicium | Si | 14 | 28,0855 |
| Stickstoff | N | 7 | 14,00674 |
| Strontium | Sr | 38 | 87,62 |
| Tellur | Te | 52 | 127,60 |
| Thallium | Tl | 81 | 204,3833 |
| Wasserstoff | H | 1 | 1,00794 |
| Xenon | Xe | 54 | 131,29 |
| Zinn | Sn | 50 | 118,710 |
| Cadmium | Cd | 48 | 112,411 |
| Chrom | Cr | 24 | 51,9961 |
| Cobalt | Co | 27 | 58,933200 |
| Eisen | Fe | 26 | 55,845 |
| Gold | Au | 79 | 196,96655 |
| Hafnium | Hf | 72 | 178,49 |
| Iridium | Ir | 77 | 192,217 |
| Kupfer | Cu | 29 | 63,546 |
| Lanthan | La | 57 | 138,9055 |
| Mangan | Mn | 25 | 54,938049 |
| Molybdän | Mo | 42 | 95,94 |
| Nickel | Ni | 28 | 58,6934 |
| Niob | Nb | 41 | 92,90638 |
| Osmium | Os | 76 | 190,23 |
| Palladium | Pd | 46 | 106,42 |
| Platin | Pt | 78 | 195,078 |
| Quecksilber | Hg | 80 | 200,59 |
| Rhenium | Re | 75 | 186,207 |
| Rhodium | Rh | 45 | 102,90550 |
| Ruthenium | Ru | 44 | 101,07 |
| Scandium | Sc | 21 | 44,955910 |
| Silber | Ag | 47 | 107,8682 |
| Tantal | Ta | 73 | 180,9479 |
| Titan | Ti | 22 | 47,867 |
| Vanadium | V | 23 | 50,9415 |
| Wolfram | W | 74 | 183,84 |
| Yttrium | Y | 39 | 88,90585 |
| Zink | Zn | 30 | 65,39 |
| Zirkonium | Zr | 40 | 91,224 |
| Cer | Ce | 58 | 140,116 |
| Dysprosium | Dy | 66 | 162,50 |
| Erbium | Er | 68 | 167,26 |
| Europium | Eu | 63 | 151,964 |
| Gadolinium | Gd | 64 | 157,25 |
| Holmium | Ho | 67 | 164,93032 |
| Lutetium | Lu | 71 | 174,967 |
| Neodym | Nd | 60 | 144,24 |
| Praseodym | Pr | 59 | 140,90765 |
| Samarium | Sm | 62 | 150,36 |
| Terbium | Tb | 65 | 158,92534 |
| Thorium | Th | 90 | 232,0381 |
| Thulium | Tu | 69 | 168,93421 |
| Uran | U | 92 | 238,0289 |
| Ytterbium | Yb | 70 | 173,04 |
In einer auf Aktivitäten anstatt Konzentrationen beruhenden Skala sind die pKS-Werte des Oxonium- und des Hydroxid-Ions 0 und 14 anstatt −1,7 und 15,7.
| HA | A− | pKS |
|---|---|---|
| HClO4 | ClO4− | ≈−9 |
| HCl | Cl− | ≈−6 |
| H2SO4 | HSO4− | ≈−3 |
| H3O+ | H2O | −1,7 |
| HNO3 | NO3− | −1,3 |
| HSO4− | SO42− | 1,9 |
| H2SO3 | HSO3− | 1,9 |
| H3PO4 | H2PO4− | 2,1 |
| [Fe(H2O)6]3+ | [Fe(H2O)5(OH)]2+ | 2,4 |
| CH3COOH | CH3COO− | 4,7 |
| [Al(H2O)6]3+ | [Al(H2O)5(OH)]2+ | 5,0 |
| (H2CO3) | HCO3− | 6,4 |
| H2S | HS− | 7,0 |
| H2PO4− | HPO42− | 7,2 |
| NH4+ | NH3 | 9,3 |
| [Zn(H2O)5–6]2+ | [Zn(H2O)4–5(OH)]+ | 9,5 |
| [Ni(H2O)6]2+ | [Ni(H2O)5(OH)]+ | 10 |
| HCO3− | CO32− | 10,3 |
| HPO42− | PO43− | 12,0 |
| HS− | S2− | 14,0 |
| H2O | OH− | 15,7 |
| C2H5OH | C2H5O− | ≈18 |
| NH3 | NH2− | ≈23 |
| OH− | O2− | ≈24 |
| ox. | red. | n e− | E°/V |
|---|---|---|---|
| Li+ | Li | 1 | −3,05 |
| Ca2+ | Ca | 2 | −2,87 |
| Na+ | Na | 1 | −2,71 |
| Al3+ | Al | 3 | −1,69 |
| Zn2+ | Zn | 2 | −0,76 |
| Fe2+ | Fe | 2 | −0,41 |
| Ni2+ | Ni | 2 | −0,26 |
| Sn2+ | Sn | 2 | −0,14 |
| Pb2+ | Pb | 2 | −0,13 |
| 2 H3O+ | H2 + 2 H2O | 2 | ±0 |
| Sn4+ | Sn2+ | 2 | +0,15 |
| Cu2+ | Cu+ | 1 | +0,16 |
| Cu2+ | Cu | 2 | +0,35 |
| I2 | 2 I− | 2 | +0,54 |
| Fe3+ | Fe2+ | 1 | +0,77 |
| Ag+ | Ag | 1 | +0,80 |
| O2 + 2 H3O+ | H2O2 + 2 H2O | 2 | +0,68 |
| Hg2+ | Hg | 2 | +0,86 |
| NO3− + 4 H3O+ | NO + 6 H2O | 3 | +0,96 |
| O2 + 4 H3O+ | 6 H2O | 4 | +1,23 |
| MnO2 + 4 H3O+ | Mn2+ + 6 H2O | 2 | +1,23 |
| Cl2 | 2 Cl− | 2 | +1,36 |
| Au3+ | Au | 3 | +1,42 |
| PbO2 + SO42− + 4 H3O+ | PbSO4 + 6 H2O | 2 | +1,68 |
{kind=link}
{kind=link}